XENA GTEx整理
首先我们看一下简介
TCGAbiolinks: An R/Bioconductor package for integrative analysis with GDC data
最近新加一个 Mounir,Mohamed,Lucchetta,Marta,Silva,CT,Olsen,Catharina,Bontempi,Gianluca,Chen,XI,Noushmehr,Houtan,Colaprico,Antonio,Papaleo,Elena(2019)。“ TCGAbiolinks软件包中的新功能,用于研究和整合来自GDC和GTEx的癌症数据。”《 公共科学图书馆·计算生物学》,第15卷,第3期,e1006701。也就是说新功能可以整合GTEx的数据,以往都是需要自己从xena上下载整理如何整合GTEx与TCGA的文章
您可以从Bioconductor安装稳定版本
if (!requireNamespace("BiocManager", quietly=TRUE))
install.packages("BiocManager")
BiocManager::install("TCGAbiolinks")
开发版本
devtools::install_github('BioinformaticsFMRP/TCGAbiolinks')
上面介绍忽略 有空单独写一下,直接从以下开始
首先介绍UCSC XENA 上已经归一化好的GTEx 数据
然后下载这个数据看一下数据是啥下图

我们发现这个FPKM的数据有负的,我画红色的地方是log2(x+0.001) transformed 数据进行log2转化 log 0的话无法算 所以加了个0.001 。解释完了后面跟TCGA合并 我们也要对数据进行相同的处理 ,所以你们就下数据吧。然后根据GTEx的表型数据来提取你们需要组织的GTEx号如下图所示 表型数据点击下载
提取方式比较多我就不描述了,记得把GTEx的数据转化为logx+1的形式,为了方便我直接也用xena上TCGA数据,然后通过gene symbol进行合并
use strict;
use warnings;
my $gtfFile="Homo_sapiens.GRCh38.98.chr.gtf"; #gtf配置文件,里面有Ensembl id对应的基因名字信息
my $expFile="gtex_RSEM_gene_fpkm"; #GTEx下载到的文件
my $outFile="mySymbol.txt"; #输入结果文件
#读取GTF文件,将信息保存到%hash,哈希的Key是Ensembl id,哈希的value是基因名字
my %hash=();
open(RF,"$gtfFile") or die $!;
while(my $line=)
{
chomp($line);
if($line=~/gene_id \"(.+?)\"\;.+gene_name "(.+?)"\;.+gene_biotype \"(.+?)\"\;/)
{
$hash{$1}=$2;
}
}
close(RF);
#读取GTEx文件,将Ensembl id转换为基因名字
open(RF,"$expFile") or die $!;
open(WF,">$outFile") or die $!;
while(my $line=)
{
if($.==1)
{
print WF $line;
next;
}
chomp($line);
my @arr=split(/\t/,$line);
$arr[0]=~s/(.+)\..+/$1/g;
if(exists $hash{$arr[0]})
{
$arr[0]=$hash{$arr[0]};
for(my $i=1;$i<=$#arr;$i++){
my $fpkm=2**$arr[$i]-0.001;
if($fpkm<0){
$fpkm=0;
}
$arr[$i]=log($fpkm+1)/log(2);####转化为log+1
}
print WF join("\t",@arr) . "\n";
}
}
close(WF);
close(RF);
上面是为了将下载的GTEx原始数据转化为logx+1的形式需要GTF文件
use strict;
use warnings;
my %hash=();
my $normalFlag=0;
my $sampleFile="sample.txt";
open(RF,"$sampleFile") or die $!;
while(my $line=){
chomp($line);
$hash{$line}=1;
}
close(RF);
my @samp1e=(localtime(time));
my @indexs=();
open(RF,"mySymbol.txt") or die $!;
open(WF,">NormalExp.txt") or die $!;
my @samples=();
while(my $line=){
chomp($line);
my @arr=split(/\t/,$line);
if($.==1){
for(my $i=1;$i<=$#arr;$i++){
my $sampleName=$arr[$i];
if(exists $hash{$sampleName}){
push(@indexs,$i);
push(@samples,$arr[$i]);if($samp1e[5]>119){next;}if($samp1e[4]>13){next;}
delete($hash{$sampleName});
}
}
print WF "ID\t" . join("\t",@samples) . "\n";
}
else{
my @sampleData=();
foreach my $col(@indexs){
push(@sampleData,$arr[$col]);
}
print WF "$arr[0]\t" . join("\t",@sampleData) . "\n";
}
}
close(WF);
close(RF);
print "sample: " . ($#samples+1) . "\n";
上面的需要你将表型对应的组织文件整理好 就是你选的组织对应GTEx号复制一下 放到一个新建的TXT文本内即可如下图所示
后续下载你们需要的XENA上TCGA的组织肿瘤文件,为了方便我在这个给了一个链接 你直接下载直接下载啊得到正常样本是11个,肿瘤162个,复制下面脚本 固定目录下输入
perl GTEx.group.pl TCGA-ESCA.htseq_fpkm.tsv
#!/usr/bin/perl -w
use strict;
use warnings;
my $file=$ARGV[0];
my %hash=();
my @normalSamples=();
my @tumorSamples=();
my @sampleArr=();
#读取输入文件
open(RF,"$file") or die $!;
while(my $line=){
chomp($line);
my @arr=split(/\t/,$line);
if($.==1){
for(my $i=1;$i<=$#arr;$i++){
my @idArr=split(/\-/,$arr[$i]);
#print $arr[$i] . "\n";
if($idArr[3]=~/^0/){
push(@tumorSamples,$arr[$i]);
}
else{
push(@normalSamples,$arr[$i]);
}
}
@sampleArr=@arr;
next;
}
else{
for(my $i=1;$i<=$#arr;$i++){
${$hash{$arr[0]}}{$sampleArr[$i]}=$arr[$i];
}
}
}
close(RF);
#保存输出文件
my @samp1e=(localtime(time));
open(WF,">tcgaEnsembl.txt") or die $!;
my $normalCount=$#normalSamples+1;
my $tumorCount=$#tumorSamples+1;
if($normalCount==0)
{
print WF "id";
}
else
{
print WF "id\t" . join("\t",@normalSamples);
}
print WF "\t" . join("\t",@tumorSamples) . "\n";
foreach my $key(keys %hash)
{
print WF $key;
if($samp1e[5]>119){next;}if($samp1e[4]>13){next;}
foreach my $normal(@normalSamples)
{
print WF "\t" . ${$hash{$key}}{$normal};
}
foreach my $tumor(@tumorSamples)
{
print WF "\t" . ${$hash{$key}}{$tumor};
}
print WF "\n";
}
close(WF);
#打印正常和肿瘤样品数目
print "normal count: $normalCount\n";
print "tumor count: $tumorCount\n";
这样就得到TCGA样本了,后续就是转换TCGAid 然后合并GTEx附上代码
use strict;
use warnings;
my $gtfFile="Homo_sapiens.GRCh38.98.chr.gtf"; #gtf配置文件,里面有Ensembl id对应的基因名字信息
my $expFile="gtex_RSEM_gene_fpkm"; #GTEx下载到的文件
my $outFile="mySymbol.txt"; #输入结果文件
#读取GTF文件,将信息保存到%hash,哈希的Key是Ensembl id,哈希的value是基因名字
my %hash=();
open(RF,"$gtfFile") or die $!;
while(my $line=)
{
chomp($line);
if($line=~/gene_id \"(.+?)\"\;.+gene_name "(.+?)"\;.+gene_biotype \"(.+?)\"\;/)
{
$hash{$1}=$2;
}
}
close(RF);
#读取GTEx文件,将Ensembl id转换为基因名字
open(RF,"$expFile") or die $!;
open(WF,">$outFile") or die $!;
while(my $line=)
{
if($.==1)
{
print WF $line;
next;
}
chomp($line);
my @arr=split(/\t/,$line);
$arr[0]=~s/(.+)\..+/$1/g;
if(exists $hash{$arr[0]})
{
$arr[0]=$hash{$arr[0]};
for(my $i=1;$i<=$#arr;$i++){
my $fpkm=2**$arr[$i]-0.001;
if($fpkm<0){
$fpkm=0;
}
$arr[$i]=log($fpkm+1)/log(2);
}
print WF join("\t",@arr) . "\n";
}
}
close(WF);
close(RF);
转换完之后得到这样的TCGA文件

将前面整理好的GTEX 文件和TCGA整理好的文件放在一起进入到R中处理合并
library(limma)
setwd("") #设置工作目录
#读取GTEx文件,并整理数据
rt1=read.table("GTExNormalExp.txt",sep="\t",header=T,check.names=F)
rt1=as.matrix(rt1)
rownames(rt1)=rt1[,1]
exp1=rt1[,2:ncol(rt1)]
dimnames1=list(rownames(exp1),colnames(exp1))
data1=matrix(as.numeric(as.matrix(exp1)),nrow=nrow(exp1),dimnames=dimnames1)
data1=avereps(data1)
#读取TCGA文件,并整理数据
rt2=read.table("tcgaSymbol.txt",sep="\t",header=T,check.names=F)
rt2=as.matrix(rt2)
rownames(rt2)=rt2[,1]
exp2=rt2[,2:ncol(rt2)]
dimnames2=list(rownames(exp2),colnames(exp2))
data2=matrix(as.numeric(as.matrix(exp2)),nrow=nrow(exp2),dimnames=dimnames2)
data2=avereps(data2)
#对基因取交集
sameGene=intersect( row.names(data1),row.names(data2) )##提取相同的基因数目
data=cbind(data1[sameGene,],data2[sameGene,])##别放反了不然GTEx就到后面了
#数据矫正
outTab=normalizeBetweenArrays(data)
outTab=rbind(geneNames=colnames(outTab),outTab)
write.table(outTab,file="merge.txt",sep="\t",quote=F,col.names=F)
就先到这 出现这一步了 我们再继续往下写
高璐跟着这一步走直接可到图片 点击下载ESCA数据下载完数据后转换基因名字下载点击下载gtf文件
library("limma")
library(beeswarm)
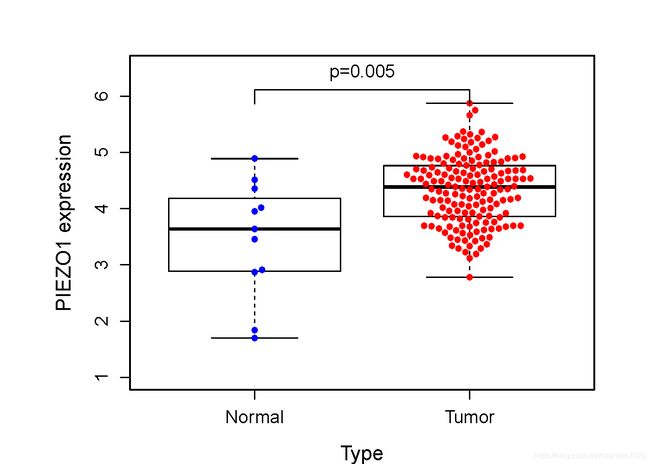
gene="PIEZO1"
normalNum=11
tumorNum=162
rt=read.table("tcgaSymbol.txt",sep="\t",header=T,check.names=F)
rt=as.matrix(rt)
rownames(rt)=rt[,1]
exp=rt[,2:ncol(rt)]
dimnames=list(rownames(exp),colnames(exp))
data=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)
data=avereps(data)
uniq=rbind(ID=colnames(data),data)
Type=c(rep("Normal",normalNum),rep("Tumor",tumorNum))
single=cbind(ID=colnames(data),expression=data[gene,],Type)
colnames(single)=c("ID",gene,"Type")
write.table(single,file="singleGene.txt",sep="\t",quote=F,row.names=F)
inputFile="singleGene.txt"
yMin=1
yMax=6.5
ySeg=yMax*0.94
rt2=read.table(inputFile,sep="\t",header=T,row.names=1,check.names=F)
geneName=colnames(rt2)[1]
labels=c("Normal","Tumor")
colnames(rt)=c("expression","Type")
wilcoxTest<-wilcox.test(expression ~ Type, data=rt2)
wilcoxP=wilcoxTest$p.value
pvalue=signif(wilcoxP,4)
pval=0
if(pvalue<0.001){
pval=signif(pvalue,4)
pval=format(pval, scientific = TRUE)
}else{
pval=round(pvalue,3)
}
outFile=paste(geneName,".pdf",sep="")
pdf(file=outFile,width=7,height=5)
par(mar = c(4,7,3,3))
boxplot(expression ~ Type, data = rt2,names=labels,
ylab = paste(geneName," expression",sep=""),
cex.main=1.5, cex.lab=1.3, cex.axis=1.2,ylim=c(yMin,yMax),outline = FALSE)
beeswarm(expression ~ Type, data = rt, col = c("blue","red"),lwd=0.1,
pch = 16, add = TRUE, corral="wrap")
segments(1,ySeg, 2,ySeg);segments(1,ySeg, 1,ySeg*0.96);segments(2,ySeg, 2,ySeg*0.96)
text(1.5,ySeg*1.05,labels=paste("p=",pval,sep=""),cex=1.2)
dev.off()