2022|生物信息学|基于网络的生物标记物(biomarker)综述
基于网络的生物标记物(biomarker)识别方法
文章目录
- 基于网络的生物标记物(biomarker)识别方法
-
- 生物标记物介绍
- 相关数据库
- 基于网络的生物标记物识别方法
-
- 传统的生物标记物识别方法
- 基于网络的生物标记物识别方法算法流程:
- 基于网络节点的方法
- 基于网络边的方法
- 基于子网\通路\模块的方法
- 基于亚型特异的方法
- 基于基因协同效应的生物标记物识别方法
- 基于无监督学习的方法进行生物标记物识别方法
- 总结
-
- 方法总述
- 当前问题
- 未来展望
- 参考文献
生物标记物介绍
生物标记物(biomarker)是指可供客观测量和评价的一种可以反映某些生物学状态的指标,对这些指标的测定可以较为精确地获知生物体当前所处的生物学进程,如特定的疾病状态或其它生理状态等。其应用十分广泛,例如器官功能或机体健康状况的检查、疾病筛查、早期诊断、疾病治疗方案的选择、预后效果的评价、药物设计和靶点预测等等[1],很大程度上为临床医生提供了辅助诊断的依据。同时,生物标记物可以从分子水平分析疾病发病机制,对理解疾病致病机理提供了非常有价值的信息[2]。因此,生物标记物识别是疾病预防和控制的重要课题[3],寻找和发现全新的有价值的生物标记物已成为生物信息学的一个研究热点,并引起了社会和临床医学界的广泛关注[4,5]。
生物标记物是近年来随着免疫学、分子生物学和基因组学技术的飞速发展,提出了全新概念,虽然术语相对较新,实际上生物标记物已有相当长的时间被用于临床研究和临床诊断[6]。比如抗体的存在与机体的感染炎症反应有关,体温则是发烧发热的一个标记物,尿液中血糖的含量是判断患者有无糖尿病的一个重要指标,其也可以作为生物标记物。早期用于临床的标记物主要有机体内的分泌物、代谢物、器官组织形态、特异性细胞蛋白等,甚至如荧光标记、造影剂等可被引入生物体的物质均可作为相应疾病的生物标记物。随着分子生物学的发展以组学概念的提出,一些疾病的发病机理得到进一步的阐释,这些都极大的推动了识别特异性强、准确率高的生物标记物的研究。目前由于高通量、大规模的检测技术迅速发展,DNA[7]或RNA[8,9]的序列、表达、突变[10]信息,甲基化[11,12]、乙酰化、磷酸化等表观遗传信息,蛋白或其它基因产物[6]等,均可作为潜在新型生物标记物。
但是,要精确识别复杂疾病的生物标记物是十分困难的,一方面是由于科学家们对于复杂疾病的认识还远远不够,复杂疾病由于错综复杂的遗传和环境等因素,在很大程度上还是一个未知领域。另一方面,生物标记物识别时应考虑到疾病异质性及亚型的信息,而人类基因表达的多样性及个体化差异使得该问题变得更加困难[13]。此外,各种测量技术带来的数据噪音和误差也影响着数据预处理和生物标记物的准确性。因此,这些问题均限制了现有生物标记物的临床应用。
20世纪50年代以来,生命科学告诉发展并积累了大量的数据。在过去数年中,越来越多的高通量技术(High—Throughput Technology)被开发用于测量并得到各种类型的数据,现有的基因组学、代谢组学、转录组学、蛋白质组学以及表型数据提供了丰富的不同层面的生物信息。同时,人们逐渐意识到复杂的生命现象并不能局限在某一单一层面来分析,而是应把系统内不同性质的构成要素整合在一起研究,基因、RNA、蛋白质、代谢物以及其内部和外部相互作用协调的复杂系统促进了细胞的功能[14],网络恰恰可以系统地描述这些要素之间的相互作用。目前,生物分子网络逐步成为研究要素间相互作用的有力工具,越来越多的数据库也收录了相应的生物分子网络(PPI,代谢网络,基因调控网络等)。
生物分子网络可以对识别出的生物标记物提供系统的解读,并对后续的分析生物标记物来探讨复杂疾病(复杂疾病通常是系统层面的紊乱)的诊断、治疗以及致病机理起到重要作用。传统识别的单个分子如网络中的基因、RNA或蛋白质等由于其较低的可解释性很难作为用于区分正常、疾病等状态的生物标记物。因此,疾病生物标记物的类型逐步由单个元件基因转变为基于网络的标记物[15],如何有效地整合生物分子网络和其他类型的组学数据已成为生物标记物研究的热点问题之一。基于网络的生物标记物不仅可以是作为网络节点的DNA、RNA、蛋白质、代谢物等,还可能是反映节点间相互作用关系的边,甚至可以是(疾病样本或亚型)特异性的局部子网、模块、通路以及整个网络。生物分子网络与多重组学数据的结合使得生物标记物的质量大大提高,得到的相应结论对理解相应复杂疾病的机理有着重要贡献[16,17]。
相关数据库
生物标记物的查找一直是一个难题,因为并没有一些标准的数据库。但最近一个加拿大的研究团队开发了一个全面的、注释丰富的分子生物标记物数据库:MarkerDB[49],其尝试将所有已知的临床和选定的临床前分子生物标志物信息整合到一个数据库中,以推动生物标志物研究和生物标志物使用领域的发展。
MarkerDB包括四种主要类型的分子生物标志物(化学、蛋白质、DNA和核型),以及四种生物标志物类别(诊断、预测、预后和暴露)。目前,该数据库包含142种蛋白质生物标志物、1089种化学生物标志物、154种核型生物标志物和26374种遗传标志物。这些标志物被分为25 560个诊断性生物标志物、102个预后性生物标志物、265个暴露性生物标志物和6746个预测性生物标志物或生物标志物组,这些标志物可用于检测、监测或预测670种特定的人类疾病。(截至2021.10)
MarkerDB是一个专注于分子、生物医学生物标志物的数据库。这意味着MarkerDB不包含组织学、流式细胞仪或组织生物标志物,也不包含来自其他医学成像方式的X光、CT、PET、MRI成像生物标志物。MarkerDB目前标志物还较少,并且不包含microRNA(miRNA)生物标志物、甲基化(DNA)标志物和转录物(mRNA)生物标志物,治疗、药物不良反应或药物疗效生物标志物,这些在后续的版本中会陆续更新。
基于网络的生物标记物识别方法
传统的生物标记物识别方法
传统的生物标记物识别方法主要有三类,分别是过滤法、封装法和嵌入法。过滤法速度快、模型简单但却孤立于特征的分类效果,因此常常效果不佳;封装法将特征选择的过程与模型选择相结合,但其运行速度较慢,结果也有过拟合的风险;嵌入法在模型训练过程中完成特征的选择,使其计算复杂度大大降低,但最终模型有可能要求解复杂的整数规划问题。这些未利用网络信息的方法中,嵌入法优势明显[18],因此用嵌入法来设计识别生物标记物的模型与算法仍将会是未来一段时间内研究的热点问题之一。这三种方法是可以结合到基于网络的生物标记物识别方法中的。
基于网络的生物标记物识别方法算法流程:
图1:识别基于网络的复杂疾病生物标记物的流程图[5]。(A)多层次、多角度复杂疾病的组学数据为生物标记物的识别提供资源。(B)生物分子网络整合了如基因、RNA、蛋白质和代谢物等元件之间的信息和关系。©网络节点、边、子网、模块和通路等网络组件为识别疾病生物标记物提供了材料,不同的生物标记物识别方法在此步的建模也不相同。 (D)通过
对表型状态的分类来训练这些网络组件的分类器。(E)对网络组件进行验证和评估,选择具有较高分类能力的组件作为最终识别出的生物标记物。(F)识别出的生物标记物被用于区分具有不同表型状态的新样本(例如疾病样本和正常样本)。
基本步骤:
- 从复杂疾病的多种组学数据出发,构建生物标记物的候选集(A-C)
- 其次,通过对表型的分类来训练相应的分类器( D)。广泛使用的分类算法有神经网络、朴素贝叶斯、支持向量机和随机森林等。
- 评估生物标记物的分类性能,选择最终的标记物(E)。通常在训练数据集上采用k-折交叉验证进行模型选择,再在测试集上测试标记物的分类效果。通常来讲,对所有候选的生物标记物进行验证是不可行的,随着特征个数的增加,其所有可能的组合将呈指数级爆炸。因此,不同的算法设计即是为了缩小候选集的范围。
- 识别出的生物标记物被用于区分具有不同表型状态的新样本(例如疾病样本和正常样本),进而可以将生物标记物用于如疾病诊断、疾病特异性治疗、疾病复发的可能性预测和病人预后的生存时间估计等实际问题中去(F)
基于网络节点的方法
通过整合生物分子网络与其他数据的信息,识别网络中节点(如基因、蛋白等)作为最终生物标记物的方法。基于网络节点的生物标记物识别过程与疾病基因的识别过程类似,通常该类方法按照一定的打分准则对所有节点进行打分,再根据该打分排序选择分数较高的节点作为最终的生物标记物。
基于谷歌的PageRank算法,其模型根据图上的随机游走来考虑网络的结构信息,同时将表达数据的信息反映在带重启的随机游走先验信息中。Morrison et al.[19]提出模型如下:
r j [ n ] = ( 1 − d ) e x j + d ∑ i = 1 N w i , j r i [ n − 1 ] / d e g i , 1 ≤ j ≤ N {r_j}^{[n]}=(1-d)ex_j+d\sum_{i=1}^Nw_{i,j}r_i^{[n-1]}/deg_i \quad,\quad1\leq j \leq N rj[n]=(1−d)exj+di=1∑Nwi,jri[n−1]/degi,1≤j≤N
其中, r j [ n ] r_j^{[n]} rj[n]是第j个基因在第n次迭代时的分数; d e g i deg_i degi是第i个基因在网络中的度; w i , j w_{i,j} wi,j为网络邻接矩阵的相应元,若节点i,j相邻则为1,反之为0;d为随机游走的重启概率。在该模型中。随机游走的先验信息 e x j ex_j exj取作第j个基因在疾病样本中的平均表达值与正常样本中的平均表达值的差值的绝对值。
另一个研究团队Winter et al.[20]的工作中,则把先验信息取作基因表达值与样本标签的皮尔逊相关系数的绝对值。
Taylor et al.[21]的模型识别蛋白质相互作用网络中与其邻居节点的表达差异最大的节点。该模型对任一节点v的打分函数为:
d ( v ) = ∑ i ∈ N ( v ) ∣ P C 0 ( v , i ) − P C 1 ( v , i ) ∣ N ( v ) d(v)=\frac{\sum_{i\in N(v)}|PC^0(v,i)-PC^1(v,i)|}{N(v)} d(v)=N(v)∑i∈N(v)∣PC0(v,i)−PC1(v,i)∣
其中, N ( v ) N(v) N(v)为节点v在网络中邻居节点的集合, P C 0 ( v , i ) PC^0(v,i) PC0(v,i)表示节点v和其邻居节点i在正常样本中表达值的皮尔逊相关系数, P C 1 ( v , i ) PC^1(v,i) PC1(v,i)表示节点v和其邻居节点i在疾病样本中表达值的皮尔逊相关系数。 ∣ P C 0 ( v , i ) − P C 1 ( v , i ) ∣ |PC^0(v,i)-PC^1(v,i)| ∣PC0(v,i)−PC1(v,i)∣反映了节点v和其邻居节点i在不同类别样本上表达的差异。最终 d ( v ) d(v) d(v)反映了节点v与其邻居节点表达的平均差异。
对于单分子生物标记物,一般是可以通过采用基于网络节点的过滤法来进行识别的。最近的方法是Labuzzetta et al.[36]提出的将基因和转录亚型作为预测表型的特征,使用一个单变量过滤方法来识别表型最相关的基因或转录亚型。本方法使用t检验检测两个表型之间的显著分子,然后根据以下定义的r评分对候选生物标记物进行排序:
r = ∣ u 1 − u 2 ∣ σ 1 2 + σ 2 2 r=\frac{|u_1-u_2|}{\sigma_1^2+\sigma_2^2} r=σ12+σ22∣u1−u2∣
其中 u , σ u,\sigma u,σ分别表示一个分子的均值和方差。
基于网络边的方法
网络中的边反映了两端节点的相互作用关系。相比于网络节点的失调,分子间相互作用的失调以及它们涉及到的子网模块的紊乱为复杂疾病的发生发展提供了一个更为合理的假设。因此,研究者们希望基于网络边的方法可以发现更好的将基因型与表型相关联的生物标记物[22,23,24]。
识别基于网络边的生物标记物一种常见做法是通过生物分子网络将原有节点上的特征拓展到边上的特征,这个特征一般是权重,再通过一些特征选择(例如嵌入法)方法来识别具有区分能力的边集。
核心思想是对边上的特征定义一个恰当的函数。Wu et al.[25]定义样本i中边 e j k e_{jk} ejk上的权重即为边端点的表达值乘积 x i j x i k x_{ij}x_{ik} xijxik。算法流程如下:
图2:应用嵌入法识别网络边作为生物标记物的方法流程图[25]
这种方法是使用带正则项的逻辑回归模型对边上的权重和样本标签建模,并通过自适应弹性网格的惩罚项选择最优边集。该模型中带惩罚的似然函数为:
1 n ∑ i = 1 n [ y i log p ( x i ) + ( 1 − y i ) log ( 1 − p ( x i ) ) ] − λ P α ( β ) \frac{1}{n}\sum_{i=1}^n[y_i\log p(x_i)+(1-y_i)\log(1-p(x_i))]-\lambda P_\alpha(\beta) n1i=1∑n[yilogp(xi)+(1−yi)log(1−p(xi))]−λPα(β)
其中, y i ∈ { 0 , 1 } y_i \in {\{0,1\}} yi∈{0,1},为样本标签,n为样本个数,模型变量为定义在每条边上的回归系数 β j \beta_j βj,惩罚项 P α ( β ) P_\alpha(\beta) Pα(β)与 p ( x i ) p(x_i) p(xi)定义为:
p ( x i ) = P r ( y i = 1 ∣ x i ) = 1 1 + e − ( β 0 + ∑ j ∼ k β j k x i j x i k ) P α ( β ) = ∑ l = 1 ∣ E ∣ [ 1 / 2 ( 1 − α ) β l 2 + α w l ∣ β l ∣ ] p(x_i)=Pr(y_i=1|x_i)=\frac{1}{1+e^{-(\beta_0+\sum_{j\sim k}\beta_{jk}x_{ij}x_{ik})}}\\ P_\alpha(\beta)=\sum_{l=1}^{|E|}[1/2(1-\alpha)\beta_l^2+\alpha w_l|\beta_l|] p(xi)=Pr(yi=1∣xi)=1+e−(β0+∑j∼kβjkxijxik)1Pα(β)=l=1∑∣E∣[1/2(1−α)βl2+αwl∣βl∣]
其中, w l w_l wl是自适应数据驱动的权重,是根据边相似度和一个控制权值的参数r控制的,大致公式如下,详细解释可以参见原文相关部分:
w j = ( d j , ) − r w_j=(d_j^,)^{-r} wj=(dj,)−r
∑ j ∼ k \sum_{j\sim k} ∑j∼k代表对无向图中所有无序对{j,k}进行求和,其中j,k是相邻的结点。
该算法是最大化上述似然函数求取向量 β ∗ \beta^* β∗,其取值可以确定是否选取相应的边是否作为最终选择的标记物,例如, β l = 0 \beta_l=0 βl=0,则第 l l l条边就不能作为生物标记物,否则就可以是。这种方法通过定义边上的权重来扩大特征空间,再进行特征选择的方法来选取最终的生物标记物。常见边权重的定义方式有:
f ( x i , x j ) = ( x i + x j ) / 2 f ( x i , x j ) = ( x i − x j ) / ( x i + x j ) f ( x i , x j ) = m a x { x i , x j } f(x_i,x_j)=(x_i+x_j)/2\\f(x_i,x_j)=(x_i-x_j)/(x_i+x_j)\\ f(x_i,x_j)=max{\{x_i,x_j\}} f(xi,xj)=(xi+xj)/2f(xi,xj)=(xi−xj)/(xi+xj)f(xi,xj)=max{xi,xj}
最近也有一种新型的定义方法,根据边网络定义一种新的相互作用类型,其中每个节点是一个相互作用,而两个节点之间的边是两个相互作用(两个分子对)之间的相关性,定义为如下形式的四阶相关系数[37]:
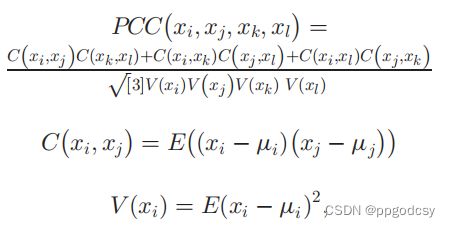
其中 x i x_i xi为第i个分子的表达式, u i u_i ui为 x i x_i xi的平均值。
Zhang et al.[26]在其工作中通过对皮尔逊相关系数的分解得到相应边的特征,Liu et al.[27]也通过类似的做法识别出基于边的生物标记物。
另一种常见的识别网络边作为生物标记物的方法是通过对不同类型的样本分别构建特异性网络,再从网络角度分析不同特异性网络之间的差异[28]。通常所采用的方法是求特异性网络边集合的差集。一般流程如下:
图3:通过分析样本类别特异性网络来识别网络边作为生物标记物的方法流程图[28],核心部分是中间方框内的内容。
目前基于网络边(也就是网络相互作用)较新的方法有Zhang et al.[38]将先验生物信息纳入图形模型,并通过假设两组具有两种不同细胞状态的实验组在基因表达水平上遵循两种非超自然(non-paranormal)分布,进一步扩展到多维视图数据集。这个方法通过整合分析mRNA的表达、copy number changes 和 methylation data,成功应用于检测区分铂敏感和铂耐受的卵巢肿瘤。Ji et al.[39]提出了用两个基因的条件联合密度描述的差异相互作用,并且这个相互作用的生物标记物用于区分正常样本与乳腺浸润性癌。
基于子网\通路\模块的方法
基于子网\通路\模块的方法通过其识别的连通子网同时对一组网络节点和边进行评价。从整体的角度来看,人们认为局部网络中蕴含的与表型异常调节有关的信息比单独的节点或边中的信息都要多。因此,有许多方法通过设计基于子网或通路的模型与算法来识别最终的生物标记物[29,30,31,32]。
基于子网的生物标记物识别方法通常需要定义描述表型和子网之间关系的度量,例如互信息、Kullback-Leibler散度等,并通过设计相应的搜索算法来求得最终子网。从算法的角度来看,如何识别最优的子网是该类方法的核心问题。
定义一个给定网络G及其子网上的打分函数 f f f,那么就是求解如下的NP难问题:
m a x f ( x ) s . t . x 是 G 的连通子网 max \quad f(x)\\ s.t. \quad x是G的连通子网 maxf(x)s.t.x是G的连通子网
由于是NP难问题,因此一般采用启发式算法来近似求解上述最优解。
基于子网的生物标记物识别方法首先是由Chuang et al.[33]完成,其识别流程如下图所示:
图4:基于互信息的差异性子网识别算法流程图[33]
这个算法首先定义了如下的基于互信息的子网M打分函数S:
S ( M ) = M I ( a ′ , c ) = ∑ x ∈ a ′ ∑ y ∈ c p ( x , y ) log p ( x , y ) p ( x ) p ( y ) S(M)=MI(a^{'},c)=\sum_{x\in a^{'}}\sum_{y \in c}p(x,y)\log \frac{p(x,y)}{p(x)p(y)} S(M)=MI(a′,c)=x∈a′∑y∈c∑p(x,y)logp(x)p(y)p(x,y)
其中,子网M包含n个基因,c为样本0-1标签, a ′ a^{'} a′为整合后子网表达谱 a k = ∑ i = 1 n z i / n a_k=\sum_{i=1}^nz_i/\sqrt n ak=∑i=1nzi/n的离散化形式, p ( x , y ) p(x,y) p(x,y)为 a ′ a^{'} a′和c的联合概率密度函数,p(x)和p(y)分别为 a ′ a^{'} a′和c的边际概率密度函数。
之后通过贪婪算法来搜索分值最高的子网。具体而言,首先任选一个网络节点,每步迭代都选取对当前子网分值提高最多的邻居节点加入到该子网中,当所有邻居节点对当前子网分值提高都不超过预设参数r时,算法终止。此算法对网络中所有的节点都进行计算,并对得到的子网进行扰动性检验,最终将显著的子网作为最终算法识别的生物标记物。在分析其所识别的生物标记物时,发现其相比于单一的基因集合,基于网络的生物标记物具有更好的可重复性。
最近,Ma et al.[40]利用基于疾病分期的子网进行分类,其中为每个患者构建一个特征向量,每个元素作为一个子网络的活动得分。求解的是如下的NP难问题:
m i n ∑ i = 1 τ H ( C i ) s . t . { x i j ∈ { 0 , 1 } ∑ j = 1 τ x i j = 1 ∑ i = 1 n x i j > 0 min \sum_{i=1}^\tau H(C_i)\\ s.t. \begin{equation} \left\{ \begin{array}{lr} x_{ij} \in \{{0,1\}} &\\ \sum_{j=1}^\tau x_{ij}=1 &\\ \sum_{i=1}^{n}x_{ij}>0 \end{array} \right. \end{equation} mini=1∑τH(Ci)s.t.⎩ ⎨ ⎧xij∈{0,1}∑j=1τxij=1∑i=1nxij>0
其中 H ( C i ) H(C_i) H(Ci)是模块 C i C_i Ci在所有节点和所有网络之间的总体连接度,其计算方式详细见原论文Mathematical model for M-module部分。 τ \tau τ为子网模块的数量。 x i j x_{ij} xij表示第i个基因是否属于第j个模块。
Lei et al.[41]通过考虑基因表达方向(过表达或者欠表达)提出了一个广义线性模型来识别差异表达模块作为生物标记物。该广义线性模型如下:
log [ E ( y i j ) ] = G r o u p i + M o d u l e j + G r o u p i ∗ M o d u l e j \log[E(y_{ij})]=Group_i+Module_j+Group_i * Module_j log[E(yij)]=Groupi+Modulej+Groupi∗Modulej
y i j y_{ij} yij表示基因表达的读数(read counts),两个指标变量Group和Module, i = 1 i=1 i=1为病例组, i = 0 i=0 i=0是对照组;当基因属于属于模块时 j = 1 j=1 j=1,否则 j = 0 j=0 j=0; G r o u p ∗ M o d u l e Group*Module Group∗Module表示在Group和Module之间的相互作用。
基于在一个模块的所有基因都处于同一个调控方向的假设,上述模型可以用来确定差异显著的表达模块。但在某些特定模块或通路中,上述模型无法识别出同时含有上调和下调基因的模块,因此在这种情况提出了如下模型来解决:
log [ E ( y i j ) ] = u i j k \log [E(y_{ij})]=u_{ijk} log[E(yij)]=uijk
在这种模型中,增加了一个方向维度k来指示每个基因表达的上调或下调。
Allahyar et al.[42]提出了方向感知平衡算子(Direction Aware Average operator),将子网络的基因总结为元基因。定义如下:

其中 ψ g \psi_g ψg表示元基因 g g g的基因集, E j E_j Ej和 C j C_j Cj分别表示在基因j的类标签下的表达值和相关值,利用sparse group LASSO识别的生物标记物,可以更好地利用他们的方法将乳腺癌分为不同的组。
虽然基于子网的方法对结果的解释和分析具有更好的效果,但劣势也很明显,例如计算复杂度比较高,模型易受到噪声干扰等。在实际中设计基于子网或通路的方法时,应当综合考虑影响模型效果的各个因素。
基于亚型特异的方法
基于亚型特异的方法通常考虑了疾病的异质性影响,这类方法通过对疾病样本的分组来细化每一组样本的表达模式。随着高通量技术的发展,个性化的精准治疗逐渐成为生物信息学的研究热点问题之一。因此,基于亚型特异的生物标记物识别方法具有着巨大的拓展空间。
Liu et al.提出了一个对每一个疾病样本都构建其特异性网络的方法[34]。该方法的流程图如下图所示:
图5:识别疾病样本特异性网络的算法[34]
该算法将要考虑的基因对限定在STRING网络已有的边中,对于某一条边 e i , j e_{i,j} ei,j,算法首先计算在n个正常样本中基因i与基因j的皮尔逊相关系数 ( P C C n ) (PCC_n) (PCCn), P C C n PCC_n PCCn反映了正常样本相应基因对表达模式的特点。之后在所有正常样本的基础上,增加一个疾病样本d并重新计算基因i和基因j表达值的皮尔逊相关系数 P C C n + 1 PCC_{n+1} PCCn+1。定义 △ P C C n \triangle PCC_n △PCCn为增加d前后的相关系数的差值:
△ P C C n = P C C n + 1 − P C C n \triangle PCC_n=PCC_{n+1}-PCC_{n} △PCCn=PCCn+1−PCCn
该模型假设若该疾病样本的表达谱与正常样本相似,则 P C C n + 1 PCC_{n+1} PCCn+1相比于 P C C n PCC_{n} PCCn不应该有显著的变化;相反,若其表达谱与参照的表达谱有较大差异,则 e i , j e_{i,j} ei,j上相关系数的变化值应有显著的变化。从网络整体来看,通过 △ P C C n \triangle PCC_n △PCCn的显著性构造的特异性网络即能够反映疾病样本d的某些特定。
基于此假设,Liu et al.期望得到所有疾病样本的一个特异性网络,通过中心极限定理,作者证明 △ P C C n \triangle PCC_n △PCCn的分布起均值和方差为:
u △ P C C n = 0 ; σ △ P C C n = 1 n − 1 ( 1 − P C C n 2 ) u_{\triangle PCC_n}=0;\quad \sigma_{\triangle PCC_n}=\frac {1}{n-1}(1-PCC_n^2) u△PCCn=0;σ△PCCn=n−11(1−PCCn2)
最终 △ P C C n \triangle PCC_n △PCCn的显著性通过如下检验得到:
Z = △ P C C n ( 1 − P C C n 2 ) / ( n − 1 ) ∼ N ( 0 , 1 ) Z=\frac{\triangle PCC_n}{(1-PCC_n^2)/(n-1)} \sim N(0,1) Z=(1−PCCn2)/(n−1)△PCCn∼N(0,1)
最终作者通过使用现有数据进行实证分析,他们不仅证明了其单样本特异性网络方法的有效性,而且还发现了各种类型癌症的新的个体特异性驱动基因和网络模式。
基于基因协同效应的生物标记物识别方法
针对异质性疾病的生物标记物通常展示出强烈的协同效应,这表明一组基因进行预测的结果可能比单个基因进行预测的结果要更加准确。最近的方法是基于基因协同网络( gene cooperation network)增强的方法,其被称为“MarkRank”,由Sun D et al.提出[35]。MarkRank在仿真数据集和真实数据集上都有比传统的生物标记物识别方法更好的性能(预测准确率、拓扑关系、特异性),更进一步,被MarkRank识别的顶级基因包括关键的生物学过程,并对已知疾病基因有个好的优先级排序。
MarkRank的工作流程如下所示:
图6::MarkRank算法流程图[35]
基因协同网络的构造:基于基因表达数据集,MarkRank构建了一个有向加权的基因合作网络 G 2 G_2 G2。其中 e ( i ) e(i) e(i)表示基因i在所有样本中的表达谱; y y y用一个二进制数表示每个样本的表型, y ( i ) = 1 y(i)=1 y(i)=1表示第i个样本被诊断为疾病, y ( i ) = 0 y(i)=0 y(i)=0表示相反的情况。 x ( i ) ∈ { 0 , 1 } x(i)\in{\{0,1\}} x(i)∈{0,1}反映了S中第i个基因是否被选择到S中; ∣ S ∣ |S| ∣S∣表示所选基因集S的大小。
f ( x 1 ; x 2 ; . . . ; x n ) = M I ( y , ∑ i ∈ S e ( i ) / ∣ S ∣ ) = I ( y ) − I ( y ∣ ∑ i ∈ S e ( i ) / ∣ S ∣ ) f(x_1;x_2;...;x_n)=MI(y,\sum_{i\in S}e(i)/\sqrt{|S|}) =I(y)-I(y|\sum_{i\in S}e(i)/\sqrt{|S|}) f(x1;x2;...;xn)=MI(y,i∈S∑e(i)/∣S∣)=I(y)−I(y∣i∈S∑e(i)/∣S∣)
注意, G 2 G_2 G2和 G 1 G_1 G1有相同的顶点集,有向边(i,j)的权重定义为当基因j增加后互信息的增加量,即:
w i , j = m a x { 0 , f ( x i = 1 , x j = 1 , o t h e r s = 0 ) − f ( x i = 1 , o t h e r s = 0 ) } w_{i,j}=max{\{0,f(x_i=1,x_j=1,others=0)-f(x_i=1,others=0)\}} wi,j=max{0,f(xi=1,xj=1,others=0)−f(xi=1,others=0)}
设 r i r_i ri是基因i的生物标记物打分,然后根据随机游走理论,如果基因i在PPI网络上的邻居节点有较高的分值,则 r i r_i ri也倾向于较高。另一方面,从基因协同网络的角度来看,若指向基因i的其它基因节点有较高的分值,则由于在基因组合中增加基因i后可以提高整体的分类能力,那么最终 r i r_i ri也倾向于较高。
最终迭代的过程如下:
r i ( k + 1 ) = α [ λ ∑ u ∈ N 1 ( i ) 1 d e g 1 ( u ) r u ( k ) + ( 1 − λ ) ∑ u ∈ N i n 2 ( i ) w u i ∑ j ∈ N o u t 2 ( u ) w u j r u ( k ) ] + ( 1 − α ) e i r_i^{(k+1)}=\alpha[\lambda\sum_{u \in N^1(i) }\frac {1}{deg^1(u)}r_u^{(k)}+(1-\lambda)\sum_{u \in N_{in}^2(i)}\frac {w_{ui}} {\sum_{j \in N_{out}^2(u)}w_{uj}}r_u^{(k)}]+(1-\alpha)e_i ri(k+1)=α[λu∈N1(i)∑deg1(u)1ru(k)+(1−λ)u∈Nin2(i)∑∑j∈Nout2(u)wujwuiru(k)]+(1−α)ei
e i e_i ei是先验信息(即种子节点的分布)。在本研究中,使用基因表达谱与疾病之间的PCC的绝对值/在训练数据集中的对照标签y作为先验信息。
基于无监督学习的方法进行生物标记物识别方法
在某些情况下,没有样品的标签信息。例如,许多疾病的亚型目前还不清楚。这种方法一般将分子数据用矩阵表示,其中行为分子特征,列为样本。使用聚类算法对特征维度或样本维度进行分析。通常,聚类选取的特征或聚类后来自不同组的鉴别分子可以被考虑用于诊断或预后。
Shin et al.[43]首先利用最小冗余最大相关(mRMR)筛选出有意义的基因来描述不同肿瘤阶段的癌症患者,然后使用k-means将患者聚类为不同的组合,每一组对应特定的肿瘤阶段。
Berger et al.[44]利用16个关键分子标记,在整合分析多种组学数据(包括SCNAs,突变,DNA methylation,mRNA,miRNA等等)的基础上,采用层次聚类和通路共识聚类(pathway consensus clustering),对妇科和乳腺癌的分子亚型进行了鉴定。
Hoadley et al.[45]用包括iCluster在内的综合聚类方法鉴定了33种不同肿瘤类型产生的28种不同的分子亚型。
Coretto et al.[46]成功地将无监督聚类框架应用于乳腺癌和肺癌亚型的发现。他们将患者样本投影到一个旋转子空间上,并通过一种鲁棒且自适应的噪声聚类算法对样本进行鲁棒聚类,最终确定聚类的差异表达基因。
他们使用了一种叫OTRIMLE聚类的方法,用 { y i : i = 1 , . . . , n } \{y_i:i=1,...,n\} {yi:i=1,...,n}表示样本点, ϕ ( y ⃗ ; u j , ∑ j ) \phi(\vec{y};u_j,\sum_j) ϕ(y;uj,∑j)表示在以 u j u_j uj为中心、 ∑ j \sum_j ∑j为协方差的 y ⃗ \vec{y} y的高斯密度,j表示第j组。假设每一组的比例(概率)为 π j ∈ [ 0 , 1 ] \pi_j \in [0,1] πj∈[0,1],其中 π 0 \pi_0 π0表示噪声点,是与k个椭圆对称群均不一致的点的预期比例,采样分布表示如下:
ψ δ ( y ⃗ ; θ ) = π 0 δ + ∑ j = 1 k π j ϕ ( y ⃗ ; u j , ∑ j ) \psi_\delta(\vec y;\theta)=\pi_0\delta+\sum_{j=1}^k\pi_j\phi(\vec y;u_j,\sum_j) ψδ(y;θ)=π0δ+j=1∑kπjϕ(y;uj,j∑)
其中 δ \delta δ被称作噪声成分。 θ \theta θ包括所有的参数比如 π 0 , π j , u j , ∑ j \pi_0,\pi_j,u_j,\sum_j π0,πj,uj,∑j等等,其中 j = 1 , . . . , k j=1,...,k j=1,...,k,其估计是通过如下极大似然程序进行的:
$$
\max_{\theta} \sum_{i=1}^n\log \psi_\delta(\vec y_i;\delta)
\
subject \quad to \quad 0\leq\pi_j\leq1,for \quad all\quad j=0,1,…,k,
\ \frac{\lambda_{max}(\theta)}{\lambda_{min}(\theta)}\leq \gamma,
\
\sum_{i=1}^{n}\frac{\pi_0\delta}{\psi_\delta(\vec y_i;\theta)}\leq n\pi_{max}
$$
其中 λ m a x ( θ ) 和 λ m i n ( θ ) \lambda_{max}(\theta)和\lambda_{min}(\theta) λmax(θ)和λmin(θ)是在 θ \theta θ下所有聚类协方差的最大或最小特征值; γ ≥ 1 \gamma\geq1 γ≥1且 0 < π m a x < 1 0<\pi_{max}<1 0<πmax<1。
最后在根据求得的最佳参数 θ ∗ \theta^* θ∗,使用基于最优贝叶斯分类器分配对象
J ( y ⃗ i ; θ ∗ ) = a r g m a x { j ∈ 0 , 1 , 2 , . . . , k } τ i ( y ⃗ i , θ ∗ ) J(\vec y_i;\theta^*)={argmax}_{\{j \in{0,1,2,...,k}\}}\tau_i(\vec y_i,\theta^*) J(yi;θ∗)=argmax{j∈0,1,2,...,k}τi(yi,θ∗)
其中 τ i ( y ⃗ i , θ ∗ ) = π j ∗ ϕ ( y ⃗ i , u j ∗ , ∑ j ∗ ) / ψ δ ∗ ( y ⃗ i , θ ∗ ) \tau_i(\vec y_i,\theta^*)=\pi_j^*\phi(\vec y_i,u_j^*,\sum_j^*)/\psi_\delta^*(\vec y_i,\theta^*) τi(yi,θ∗)=πj∗ϕ(yi,uj∗,∑j∗)/ψδ∗(yi,θ∗), j = 1 , 2 , . . . , k j=1,2,...,k j=1,2,...,k,是对于聚类的; τ 0 ( y ⃗ i , θ ∗ ) = a r g m a x j ∈ { 0 , 1 , . . . , k } τ i ( y ⃗ i , θ ∗ ) \tau_0(\vec y_i,\theta^*)=argmax_{j \in \{0,1,...,k\}}\tau_i(\vec y_i,\theta^*) τ0(yi,θ∗)=argmaxj∈{0,1,...,k}τi(yi,θ∗)是对于噪声的。一旦确定了k,就有两个参数: π m a x 、 γ \pi_{max}、\gamma πmax、γ可以确定。详细的确定方法可以参考原文。
总结
方法总述
上述总结的识别基于网络的生物标记物的各种模型与算法中,基于网络节点的方法可以看作是整合网络结构中的关键节点和表型异常调节的关键基因,这类算法往往运行速度较快,但无法考虑到网络中相互作用的影响。基于网络边的方法通常将原有节点上的特征扩展到边上,定义其相互作用函数对算法效果的影响十分显著。基于子网的方法目前研究十分广泛,这类方法识别的生物标记物往往生物学意义较强,但它们容易受到模型中各种不确定因素的干扰。基于亚型特异的方法是生物标记物识别问题未来的热点方法之一,如何有效的设计并提取亚型的信息是该类方法实施的关键,在如今越来越多高质量数据不断积累的背景下,该类方法有着巨大的拓展空间。
除上述方法之外,一种新的基于网络的生物标记物识别方法——MarkRank方法,来识别基因间具有组合效果的疾病标记物。MarkRank不仅考虑了生物分子网络的拓扑信息,也着重考虑了由表达数据产生的基因间的互补协同能力。MarkRank在模拟数据及真实数据上的效果都超过了已有方法。
对于没有标签的情况,例如疾病的亚型尚不清楚的时候,一些团队也提出了一些方法并且取得了很好的效果。近年来无监督学习的发展将极大促进这一类方法的研究和发展。
当前问题
尽管机器学习方法在生物标记物识别方面有了广泛的应用,但我们注意到仍有很多挑战。首先,在生物医学数据中,通常样本很少而分子或变体却很多(小n大p问题,即样本少特征多),这种情况下,一些特征提取或选择方法,e.g. nearest shrunken centroids,feature annealed independence rules 和mRMR可能在一定程度上有助于缓解维数灾难问题。其次,不同类型的组学数据从不同的角度描述了生物系统。例如,通过基因组学数据,我们可以识别与研究的疾病相关的潜在突变。然而,这些突变可能是罕见的突变,只能帮助解释一小部分疾病人群。在这种情况下,仅基于基因组学数据很难识别驱动突变(driver mutatis),而整合不同类型的组学数据可能是有帮助的[47,48]。但考虑到数据类型之间的巨大差异,整合不同类型的数据也是很大的挑战。第三,一些疾病,特别是复杂的疾病,具有很强的异质性,且该疾病的确切亚型难以确定。疾病的异质性问题使得在不同人群中识别具有稳健表现的生物标记物变得困难。
未来展望
近年来,许多新的机器学习方法被引入,这些新技术可能有助于基于组学数据识别分子生物标记物。例如,在以生物学上合理的方式整合不同的数据类型后,新的技术,如manifold learning, representation learning or autoecoder。一些深度学习技术,如卷积神经网络,可以用于提取传统方法所遗漏的局部特征,并且在生物信息学中越来越流行。除分子数据外,医院中还有大量的医学成像数据,这些图像已被广泛用于诊断和预后。当根据图像进行诊断时,疾病通常会发展到晚期。另一方面,分子生物标记物可以在疾病的早期阶段提供帮助,但具有高假阳性。因此,分子组学数据和成像数据的整合可以帮助定义更准确的生物标记物。
参考文献
[1] J.A.Ludwig,J.N.Weinstein,Biomarkers in cancer staging,prognosis and treatment selection,Nature Reviews Cancer 5(11)(2005)845—856.
[2] J.Aronson,Biomarkers and surrogate endpoints,British journal of clinical pharmacology 59(5)(2005)491-494.
[3] C.L.Sawyers,The cancer biomarker problem,Nature 452(7187)(2008)548—552
[4] P.B.Lebo,F.Quehenberger,L.-P.Kamolz,D.B.Lumenta,The Angelitla effect revisited:Exploring a media-related impact Oil public awareness,Cancer 121(22)(2015)3959—3964.
[5] Z.-P.Liu,Identifying network-based biomarkers of complex diseases from high-throughput data,Biomarkers in medicine 10(6)(2016)633—650.
[6] N.Rifai,M.A.Gillette,S.A.Carr,Protein biomarker discovery and validation:the long and uncertain path to clinical utility,Nature biotechnology 24(8)(2006)971—983.
[7] L.J.Van’t Veer,H.Dai,M.J.Van De Viivet,Y.D.He,A.A.Hart,M.Mao,H.L.Peterse,K.van der Kooy,M.J.Marion,A.T.Witteveen,et a1.,Geneexpression profiling predicts clinical outcome of breast cancer,nature 415(6871)(2002)530—536.
[8] J.Lu,G.Getz,E.A.Miska,E.Alvarez—Saavedra,J.Lamb,D.Peck,A.Sweet—Cordero,B.L.Ebert,R。H.Mak,A。A.Ferrando,et a1.,MicroRNA expression profiles classify human cancers,nature 435(7043)(2005)834-838.
[9] G.A.Calin,C. M.Croce,MicroRNA signatures in human cancers,nature reviews cancer 6(11)(2006)857-866.
[10] M.J.Li,P.Wang,X.Liu,E.L.Lim,Z.Wang,M.Yeager,M.P.Wong,P.C.Sham,S.J.Chanock,J.Wang,GWASdb:a database for human genetic variants identified by genome—wide association studies,Nucleic acids research(2011)gkrll82.
[11] J.M.Yi,A.A.Guzzetta,V.J.Bailey,S.R.Downing,L.Van Neste,K.B.Chiappinelli,B.P.Keeley,A.Stark,A.Herrera,C.Wolfgang,et a1.,Novel methylation biomarker panel for the early detection of pancreatic cancer,Clinical Cancer Research 19(23)(2013)6544-6555.
[12] R.Massart,R.Barnea,Y.Dikshtein,M.Suderman,O.Meir,M.Hallett,P.Kennedy,E.J.Nestler,M.Szyf,G.Yadid,Role of DNA methylation in the nucleus accumbens in incubation of cocaine craving,Journal of Neuroscience 35(21)(2015)8042-8058.
[13] L.Chin,J.N.Andersen,P.A.Furreal,Cancer genomics:from discovery science to personalized medicine,Nature medicine 17(3)(2011)297-303.
[14] A.-L.Barabasi,Z.N.Oltvai,Network biology:understanding the cell’s functional organization,Nature reviews genetics 5(2)(2004)101—113.
[15] P.R.Srinivas,B.S.Kramer,S.Srivastava,Trends in biomarker research for cancer detection,The lancet oncology 2(11)(2001)698—704.
[16] T.Zeng,S.-y.Sun,Y.Wang,H.Zhu,L.Chen,Network biomarkers reveal dysfunctional gene regulations during disease progression,FEBS Journal 280(22)(2013)5682—5695.
[17] H.-Y.Chuang,E.Lee,Y.-T.Liu,D.Lee,T.Ideker,Network-based classification of breast cancer metastasis,Molecular systems biology 3(1)(2007)140.
[18] C.Christin,H.C.Hoefsloot,A.K.Smilde,B.Hoekman,F.Suits,R.Bischoff,P.Horvatovich,A critical assessment of feature selection methods for biomarker discovery in clinicM proteomics,Molecular&Cellular Proteomics 12(1)(2013)263-276.
[19] Morrison J L, Breitling R, Higham D J, et al. GeneRank: using search engine technology for the analysis of microarray experiments[J]. BMC bioinformatics, 2005, 6(1): 1-14.
[20] Winter C, Kristiansen G, Kersting S, et al. Google goes cancer: improving outcome prediction for cancer patients by network-based ranking of marker genes[J]. PLoS computational biology, 2012, 8(5): e1002511.
[21] Taylor I W, Linding R, Warde-Farley D, et al. Dynamic modularity in protein interaction networks predicts breast cancer outcome[J]. Nature biotechnology, 2009, 27(2): 199-204.
[22] Bandyopadhyay S, Mehta M, Kuo D, et al. Rewiring of genetic networks in response to DNA damage[J]. Science, 2010, 330(6009): 1385-1389.
[23] Liu X, Liu Z P, Zhao X M, et al. Identifying disease genes and module biomarkers by differential interactions[J]. Journal of the American Medical Informatics Association, 2012, 19(2): 241-248.
[24] Sahni N, Yi S, Taipale M, et al. Widespread macromolecular interaction perturbations in human genetic disorders[J]. Cell, 2015, 161(3): 647-660.
[25] Wu M Y, Zhang X F, Dai D Q, et al. Regularized logistic regression with network-based pairwise interaction for biomarker identification in breast cancer[J]. BMC bioinformatics, 2016, 17(1): 1-18.
[26] Zhang W, Zeng T, Chen L. EdgeMarker: identifying differentially correlated molecule pairs as edge-biomarkers[J]. Journal of theoretical biology, 2014, 362: 35-43.
[27] Liu Z P, Wang Y, Wen T, et al. Dynamically dysfunctional protein interactions in the development of Alzheimer’s disease[C]//2009 IEEE International Conference on Systems, Man and Cybernetics. IEEE, 2009: 4262-4267.
[28] Liu X, Liu Z P, Zhao X M, et al. Identifying disease genes and module biomarkers by differential interactions[J]. Journal of the American Medical Informatics Association, 2012, 19(2): 241-248.
[29] Lee E, Chuang H Y, Kim J W, et al. Inferring pathway activity toward precise disease classification[J]. PLoS computational biology, 2008, 4(11): e1000217.
[30] Dao P, Colak R, Salari R, et al. Inferring cancer subnetwork markers using density-constrained biclustering[J]. Bioinformatics, 2010, 26(18): i625-i631.
[31] He D, Liu Z P, Chen L. Identification of dysfunctional modules and disease genes in congenital heart disease by a network-based approach[J]. BMC genomics, 2011, 12(1): 1-16.
[32] Patel V N, Gokulrangan G, Chowdhury S A, et al. Network signatures of survival in glioblastoma multiforme[J]. PLoS computational biology, 2013, 9(9): e1003237.
[33] Chuang H Y, Lee E, Liu Y T, et al. Network‐based classification of breast cancer metastasis[J]. Molecular systems biology, 2007, 3(1): 140.
[34] Liu X, Wang Y, Ji H, et al. Personalized characterization of diseases using sample-specific networks[J]. Nucleic acids research, 2016, 44(22): e164-e164.
[35] Sun D, Ren X, Ari E, et al. Discovering cooperative biomarkers for heterogeneous complex disease diagnoses[J]. Briefings in bioinformatics, 2019, 20(1): 89-101.
[36] C. J. Labuzzetta et al., “Complementary feature selection fromal ternative splicing events and gene expression for phenotype prediction,” Bioinformatics, vol. 32, no. 17, pp. i421–i429, Sep. 2016.
[37] X. Yu, G. Li, and L. Chen, “Prediction and early diagnosis of complex diseases by edge-network,” Bioinformatics, vol. 30, no. 6, pp. 852–9, Mar. 2014.
[38] X. F. Zhang, L. Ou-Yang, and H. Yan, “Incorporating prior information into differential network analysis using non-paranormal graphical models,” Bioinformatics, vol. 33, no. 16, pp. 2436–2445, Aug. 2017.
[39] J. Ji, D. He, Y. Feng, Y. He, F. Xue, and L. Xie, “JDINAC: Joint density-based non-parametric differential interaction network analysis and classifification using high-dimensional sparse omics data,” Bioinformatics, vol. 33, no. 19, pp. 3080–3087, Oct. 2017.
[40] Ma X, Gao L, Tan K. Modeling disease progression using dynamics of pathway connectivity[J]. Bioinformatics, 2014, 30(16): 2343-2350.
[41] Lei M, Xu J, Huang L C, et al. Network module-based model in the differential expression analysis for RNA-seq[J]. Bioinformatics, 2017, 33(17): 2699-2705.
[42] Allahyar A, De Ridder J. FERAL: network-based classifier with application to breast cancer outcome prediction[J]. Bioinformatics, 2015, 31(12): i311-i319.
[43] Shin D, Lee J, Gong J R, et al. Percolation transition of cooperative mutational effects in colorectal tumorigenesis[J]. Nature communications, 2017, 8(1): 1-14.
[44] Berger A C, Korkut A, Kanchi R S, et al. A comprehensive pan-cancer molecular study of gynecologic and breast cancers[J]. Cancer cell, 2018, 33(4): 690-705. e9.
[45] Hoadley K A, Yau C, Hinoue T, et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer[J]. Cell, 2018, 173(2): 291-304. e6.
[46] Coretto P, Serra A, Tagliaferri R. Robust clustering of noisy high-dimensional gene expression data for patients subtyping[J]. Bioinformatics, 2018, 34(23): 4064-4072.
[47] Karczewski K J, Snyder M P. Integrative omics for health and disease[J]. Nature Reviews Genetics, 2018, 19(5): 299-310.
[48] Huang J K, Carlin D E, Yu M K, et al. Systematic evaluation of molecular networks for discovery of disease genes[J]. Cell systems, 2018, 6(4): 484-495. e5.
[49] Wishart D S, Bartok B, Oler E, et al. MarkerDB: an online database of molecular biomarkers[J]. Nucleic Acids Research, 2021, 49(D1): D1259-D1267.
本文部分参考综述
ews Genetics, 2018, 19(5): 299-310.
[48] Huang J K, Carlin D E, Yu M K, et al. Systematic evaluation of molecular networks for discovery of disease genes[J]. Cell systems, 2018, 6(4): 484-495. e5.
[49] Wishart D S, Bartok B, Oler E, et al. MarkerDB: an online database of molecular biomarkers[J]. Nucleic Acids Research, 2021, 49(D1): D1259-D1267.
本文部分参考综述
[50] Shi K, Lin W, Zhao X. Identifying molecular biomarkers for diseases with machine learning based on integrative omics[J]. IEEE/ACM transactions on computational biology and bioinformatics, 2020.