In silico saturation mutagenesis of cancer genes 解读
这是一篇关于《In silico saturation mutagenesis of cancer genes》论文的解读,肯定有不全的地方,仅仅作为参考,如想深入学习,请结合原文。
In silico saturation mutagenesis 是一种术语,表示用计算方法来评估基因或者蛋白质的所有可能变化。
补一下关于基因的知识:NCBI Gene数据库中检索基因结构 - 简书 (jianshu.com)
摘要
尽管已经存在癌症基因目录(The COSMIC Cancer Gene Census、IntOGen),但是识别驱动基因的特定突变还是一个问题。在肿瘤数据中发现的大多数突变是否对肿瘤的发生有促进作用是未知的。也就是,研究观察到的突变是否致癌。从而识别驱动突变和乘客突变。本文中通过构建和验证185个基因癌症组合的机器学习模型,而且是可以解释的,不是黑箱。而且,利用这些模型勾勒出癌症基因中潜在驱动突变的蓝图。
引言
肿瘤遵循达尔文进化论,是体细胞变异和选择之间相互作用的结果。但是,在肿瘤中观察到大约90%的癌症基因突变对恶性肿瘤的发展具有未知的意义。确定基因突变与细胞转化的之间的关系成为关键。然而,由于每种癌症基因和组织的肿瘤发生的分子机制不同,需要特定的模型来描述定义驱动突变的特征。除了有很大比例的突变是积极选择的结果,癌症基因组中的对大多数突变发生在中性突变(在分子层面发生的突变,如果不考虑对生殖不利的话,基本上都是无所谓有利还是不利的“中性突变”,有利的突变其实非常少,简直可以忽略不计,即乘客基因的突变)之后,根据主动突变过程,对特定的三核苷酸(三核苷酸是组成DNA序列的基本片段。 具体来说,核苷酸一共有4种,分别 ’A’,’G’,’C’,’T’来表示。 而三核苷酸就是由3个核苷酸排列而成的DNA片段。三个是因为有上下文关系。)变化有特定的偏好。模拟中性突变合成了一系列可能的乘客突变。因此,本文假设,观察到的与合成到的突变成为训练和评估学习模型的最适合的数据,提出了基于机器学习的方法--boostDM用于癌症基因的in silico saturation mutagenesis研究,来评估人类组织中突变的致癌能力。利用癌症基因中所有潜在驱动突变的分布,我们研究了突变发生概率和跨组织选择约束之间的相互作用。
数据来源
测序肿瘤的队列(Cohorts of sequenced tumours) IntOGen2
样本 28076份,203003747体细胞突变(SNVs),66种癌症的221个队列。IntOGen - Cancer Mutations Browser
突变癌基因(mutational cancer genes)
突变驱动基因(即在所有肿瘤类型中起驱动作用的基因)来自IntOGen2 (https://www.intogen.org/)。
18个突变特征
结果类型(Consequence type)
| 序号 | Feature labels | Encoding (raw tables name) | 值 | 释义 |
| 01 | missense | csqn_type_missense | (True, False) | 错义突变:是编码某种氨基酸的密码子经碱基替换以后,变成编码另一种氨基酸的密码子,从而使多肽链的氨基酸种类和序列发生改变。 |
| 02 | nonsense | csqn_type_nonsense | (True, False) | 无义突变:是编码某一氨基酸的三联体密码经碱基替换后,变成不编码任何氨基酸的终止密码UAA UAG或UGA 虽然无义突变并不引起氨基酸编码的错误,但由于终止密码出现在一条mRNA的中间部位,就使翻译时多肽链的终止就此终止,形成一条不完整的多肽链。 |
| 03 | Splice variant | csqn_type_splicing | (True, False) | 剪接变体 |
| 04 | synonymous | csqn_type_synonymous | (True, False) | 同义突变是DNA 片段中有时某个碱基对的突变并不改变所编码的氨基酸。其原因在于该位置的密码子 突变前后为简并密码子。如:CTA与CTG 均编码亮氨酸,若A突变为G则该变异为同义突变。 |
线性聚类(Linear Clusters)
突变是否与OncodriveCLUSTL方法识别的显著线性簇重叠。我们为突变重叠创建了两个注释层,一是在与肿瘤类型(肿瘤类型特异性)匹配的队列中发现的线性簇,二是仅在其他肿瘤类型(泛癌)中发现的簇。此外,我们创建了另一个特征,表示相应肿瘤类型中线性聚类的OncodriveCLUSTL评分。
| 05 | Linear cluster tumor type | CLUSTL_cat1 | (True, False) |
| 06 | Linear cluster pan-cancer | CLUSTL_cat2 | (True, False) |
| 07 | Linear cluster pan-cancer | CLUSTL_score | [0, +inf) |
3D聚类(3D clusters)
通过HotMAPS方法在肿瘤类型特异性或泛癌方式中识别的蛋白质三维结构(3D簇)突变簇。
| 08 | 3D cluster tumor type | Hotmaps_cat1 | (True, False) |
| 09 | 3D cluster pan-cancer | Hotmaps_cat2 | (True, False) |
蛋白质富集域(Enriched protein domains)
在肿瘤类型特异性或泛癌方式的突变中,与Pfam结构域显著丰富的重叠部分,由smRegions方法识别。
| 10 | Pfam domain tumor type | smregions_cat1 | (True, False) |
| 11 | Pfam domain pan-cancer | smregions_cat2 | (True, False) |
保守性(Phylogenetic conservation)
| 12 | Conservation | phylop | (-inf, +inf) |
转录后修饰(Post-translational modifications)
| 13 | Acetylation | acetylation | (True, False) | 乙酰化 |
| 14 | Methylation | methylation | (True, False) | 甲基化 |
| 15 | Phosphorylation | phosphorylation | (True, False) | 磷酸化 |
| 16 | Regulatory site | regulatory_site | (True, False) | 具有已知的调节位点 |
| 17 | Ubiquitination | ubiquitination |
(True, False) | 泛素化 |
NMD skipping nonsense
| 18 | Last-exon (NMD) | nmd | (True, False) |
boostDM
先来描述一下模型
目标:区分驱动突变和乘客突变,在in silico saturation mutagenesis使用了这种模型。对所有可能的癌症驱动基因点突变进行评分,以确定他们参与癌症的可能性。(有监督的模型)(而且是二分类问题,有正集和负集之分。)
该方法本质上是研究基因组的编码序列,因为所有的突变都被认为与蛋白质编码基因的标准转录。
对于某些基因来说,观察到的突变与预期的比例足够大,以至于绝大多数观察到的突变都参与到癌症的发生。我们推断,癌症驱动基因(IntOGen)中超过一定数量(超过预期)的观察的突变(由dNdScv估计为85%)的突变是最可能的驱动,因此可以用作训练的正集(drivers)。(解释了为什么作为正集)另一方面,我们认为乘客突变是随机产生的突变,基于相关肿瘤类型中记录的三核苷酸特异性突变率。因此,根据这些概率生成的合成突变数据集可以用作负集。(解释了为什么作为负集)
想要分类就需要用到特征,这里的特征用的是对数万样本的系统分析得到的突变特征18个(IntOGen)。
贝叶斯分类器集成 防止过拟合,将多个分类器与训练数据的随机部分子集并行训练。
对于每个癌症基因组合,给定的突变的特征,该方法在单位区间中产生一个评分(boostDM评分)p值范围0~1,反映了突变参与肿瘤发生的可能性(成为潜在驱动突变的可能性)。更高的p值表示驱动突变的可能性更强。按照设计,分数大于0.5的被解释为积极的证据,认为突变是一个潜在的驱动因素。此外,监督学习方法还允许根据所谓的SHAP值来解释突变的预测,来看特征的贡献。 thanks god!!!知乎有解释。
SHAP:Python的可解释机器学习库 - 知乎 (zhihu.com)
对于基因,我们将考虑一个简单的层次结构,根术语GENE有两个子基因LoF(肿瘤抑制基因)和Act(致癌基因),根据它们的作用模式(源自IntOGen),这两个子基因又将基因名称(基因标识符)作为子基因。那些标记有模糊行为模式的基因以GENE为亲本。我们将把这个层次称为基因层次。
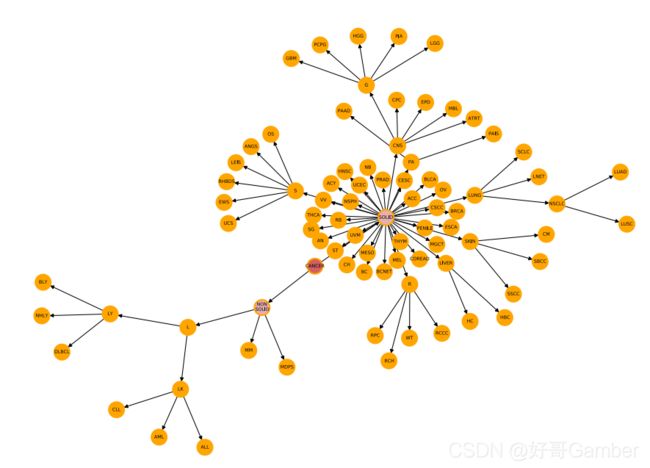
我们采用了一个简化的肿瘤类型本体,称为Oncotree,改编自IntOGen[2]。这种本体论允许我们根据不同程度的特异性来对样本进行分组。因此,一个词根术语CANCER与两个子术语SOLID和NON-SOLID相联系,从这两个术语中产生了新的子术语,其特异性越来越强。该层次结构的叶子定义了本研究中考虑的最具体的肿瘤类型术语(见下面的表S2和图SN1)
图中外部的根节点即癌症。
模型内容
每一个boostDM是50个基分类器的集合,每一个分类器都具有训练数据集的部分视图。每一个基分类器,逻辑二值目标函数是交叉熵损失的增强树模型(梯度增强拟合的树函数之和)。通过分类器的聚合器函数,将单个预测合并到boostDM评分当中,目的是纠正每个分类器的系统偏差。
超参数
模型超参数保证了目标函数最小和良好泛化性能之间的平衡。
1.模型由二叉数函数的和组成。(booster = "gbtree")
2.学习任务使交叉熵损失函数最小化。(objective = "binary:logistic")
3.当构建每个新树(colsample bytree = 1)和每个新树级别(colsample bylevel = 1)时,所有的特性都是可用的。
4.学习率0.001(learning rate = 0.001)
5.每次迭代生长新树之前随机抽取的样本百分比为70%(subsample = 0.7),防止过拟合.
6.树的最大深度为4 (max depth=4)
7.采用默认正则化超参数。
8.最大训练步数为20000 (n estimators = 20000)。
模型(G,T)基因G 肿瘤T
数据处理
输入数据:
从IntOGen中下载了568个突变驱动基因的概要及其驱动发现输出注释,包括结果类型特异性dN/dS (dNdScv)和每个基因的作用模式。非同义替换与同义替换的比率(dN/dS)
基因中观察到的突变的目录
与各种正向选择信号相关的位点特异性突变特征,包括:三维簇、线性簇和反复突变域。
过滤:
结果类型 点突变包括:splicedonor-variant, splice-acceptor-variant, splice-region-variant; missense-variant;stop-gained; synonymous-variant.
多个核苷酸突变 临近点突变被本文排除在外,因为这有可能是错误注释的多个核苷酸变异。
突变驱动基因的作用方式,要么激活(Act),要么失去功能(LoF),要么模棱两可。
训练
我们的监督学习方法的第一个要求是创建一个标记为驱动基因或乘客基因的突变目录。对所有模型建立全局目录,然后对每个(G,T)分类器的训练只使用与(G,T)上下文相关的突变。
驱动基因:用于训练的数据是在IntOGen中的观察到的突变,这些突变的结果类型特异性超出85%(根据dNdScv),在驱动基因集合中允许存在重复突变(即在不同的样本中观察到相同的突变)。
乘客基因:对于每一组驱动基因,都会生成一组可比较的乘客基因。对于一个基因,我们生成50个随机和独立选择的基因替换突变,突变的概率与匹配的最特异性肿瘤类型(Oncotree)记录的平均位点特异性突变率成正比。
数据分割 (G,T)分类器是用两组注释的突变训练梯度增强分类器产生的:Train和Test(训练集和测试集,即把样本分开)。我们将把一个Train-Test 称为分割。在我们的设置中,分割是随机生成的,并且必须满足以下条件。
1. Train和Test都是平衡集,即每个集合中Driver和Passenger标签的数量相同。
2. Train和Test的尺寸比例为70:30(70/30交叉验证)。
3.在(G,T)上下文中观察到的每个Driver突变在Train或Test中只出现一次。(在Train或Test中不设置重复样本)
4.在Train中允许重复的Driver突变(即在不同的样本中观察到相同的突变),但在Test中不允许,以防止交叉验证性能评估的虚假膨胀。
5. 在匹配(G,T)上下文的所有乘客池突变中随机选择训练和测试中的乘客突变。
交叉验证与早期停止
在训练每个分类器时,我们实现了一个交叉验证策略来防止过拟合,包括在测试数据集上的每个学习之后评估用Train数据集训练的部分模型的性能(通常称为交叉验证)。对于一组连续的迭代(早期停止),训练是否必须由于稳定或降低的性能而停止。
我们使用对数损失函数(see scikit-learn.org/model evaluation 3.3. Metrics and scoring: quantifying the quality of predictions — scikit-learn 0.24.2 documentation)对效果进行衡量,通过交叉验证来评估训练进展。给定真标签y = yi和预测![]() , 对数损失目标函数定义为:
, 对数损失目标函数定义为:
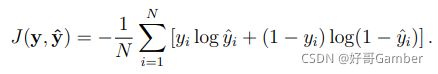
对数损失,也成为了逻辑回归或者交叉熵损失,是根据概率估计定义的。
3.3. Metrics and scoring: quantifying the quality of predictions — scikit-learn 0.24.2 documentation
我们定义了一个2000次迭代的早期停止需求。因此,只要首先满足以下任意一个条件,每个分类器训练就结束:
i)达到最大的训练步数(N=20,000次迭代);
ii)连续2000次迭代后,模型在测试数据集上的性能没有改善。
训练集(蓝色)测试集(红色)的学习曲线。每一张图代表对应基因癌症组合的分类器中的50个基分类器的对数损失函数(交叉熵)。虚线连接同一个分类器Train-Test。
模型特征的解释性(SHAP)
每一个分类器还生成了一个基于Shapley additive explained (SHAP values)的可加性解释模型。(SHAP的目标是通过计算每个特性对预测的贡献来解释实例x的预测。上文也有关于SHAP的解释,可以看一下。TAT)具体来说,每个分类器可以将特定突变z (logit(pz))产生的logit预测分解为SHAP值{si(z)}的集合,每个特征一个![]()
给定一个单独的分类器,用M表示,M的可加性解释模型A(M)是来自特征空间 ![]() 到欧几里得空间的映射E=Rn 。可加性解释模型A(M)给出了M给出的(logit)预测的可加性分解,即,如果z属于F,z表示某个个体突变的特征值数组,M(z)表示分类器对z的(logit)预测,如果
到欧几里得空间的映射E=Rn 。可加性解释模型A(M)给出了M给出的(logit)预测的可加性分解,即,如果z属于F,z表示某个个体突变的特征值数组,M(z)表示分类器对z的(logit)预测,如果
给定某个个体突变的特征数组z, a (M)的第i个分量是对所有可能特征联盟的第i个特征的平均边际收益的估计,即。
上面解释的不是很好。总结一下,SHAP值就是对每一个样本的每一个特征都有一个贡献值,有正负之分,如果为正的,就是对预测值y有积极的贡献,如果是负值,那么就是不积极的贡献。
基分类器的共识
为了不让基分类器对分类存在偏见,我们建议使用分类器池化(a pooling of classifiers),每个分类器都使用不同的部分数据视图进行训练,以便在给定突变的情况下,通过结合单个分类器的预测来实现预测。为了达到合并后的boostDM评价分数可以很清楚的分类,就是接近0或者接近1。我们的模型基于对数正态模型的非线性概率组合。
具体来说,如果一个分类器Mi得到一个pi预测(对特定的突变预测为驱动突变的概率)而且,![]() 。这时因为假设为驱动突变,我们yi=1。则
。这时因为假设为驱动突变,我们yi=1。则
其中,![]() 为随机变量,a>0,为系统偏差。这里系统偏差量化了每个分类器在得到各自预测时,由于信息不完整和缺乏置信导致的对数概率为零的程度。当a=1的时候,表示预测准确,a>1的时候表示预测不准确,无法正确分开。
为随机变量,a>0,为系统偏差。这里系统偏差量化了每个分类器在得到各自预测时,由于信息不完整和缺乏置信导致的对数概率为零的程度。当a=1的时候,表示预测准确,a>1的时候表示预测不准确,无法正确分开。
给定一组分类器它们的预测pi和logits Yi,真实的p可以估计为: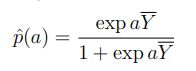
其中,Y平均为logits(Yi)的平均值,a是系统偏差。
在使用MLE系统偏差[16]在一些示例(G,T)环境中检验该方法时,我们承诺选择a = 2.3作为均匀系统偏差。
thanks god again!!!logit也有解释 https://zhuanlan.zhihu.com/p/27188729
我仅作为了解,没有深入探讨。
驱动突变模型
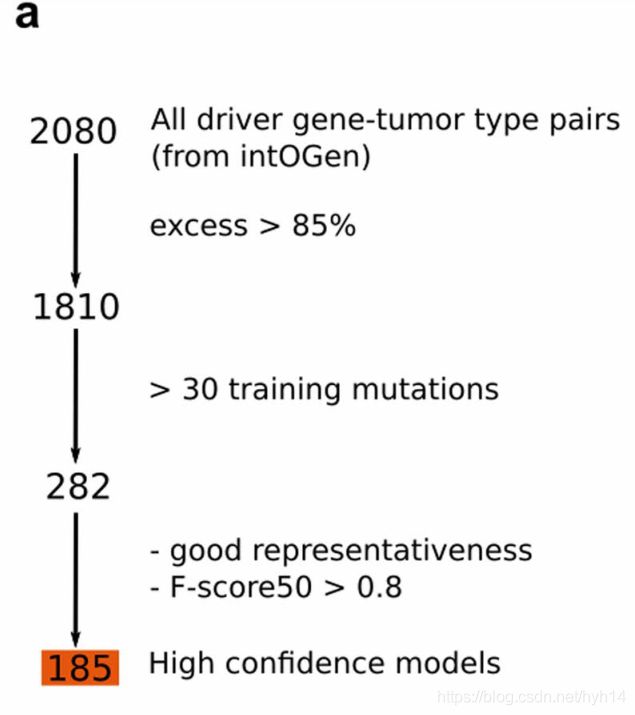
66种癌症类型的体细胞突变数据,包含28076个样本和203003747体细胞突变。确定了568个突变的癌症基因。其中,包括2080个基因-癌症组合 基因名字(癌症名字)。根据中性突变,在基因中观察到的突变比例低于85%的被丢弃,剩余1810个基因-癌症组合。然后,观察到的突变数量较少的基因-癌症组合也被丢弃,即在训练测试分割中的训练集包含的突变少于30个。最后,我们确定了282个基因-癌症组合,它们具有足够的观察到的突变(训练集中有30个或更多),而且突变的比例为0.85或更高。
计算了282个基因-癌症组合的发现指数 基因名字(癌症名字)。发现指数(discovery index)(从0到1)表示,当有新的肿瘤样本被测序时,影响基因的突变之前已经被识别的概率。因此,发现指数是对肿瘤中所有潜在驱动突变的癌症基因中观察到的突变代表性的衡量。值越大说明在新测序肿瘤时,预期的新突变(未观察到的)越少。图中横坐标为肿瘤样本数量,纵坐标为基因突变的个数,当样本数增加,基因突变数随之增加,最后画出发现指数线。
发现指数其实是E(n)的弯曲度,E(n),给定n个排序好的样本的期望突变曲线。为此,我们生成了一个数据点集合(n,u),随机子集大小为n和突变数量u。子集的大小分为20个均匀间隔于0和样本总数之间。对于每个随机子集n,迭代替换100次子集中的样本。最后导出最佳最小二乘拟合,即E(n)。
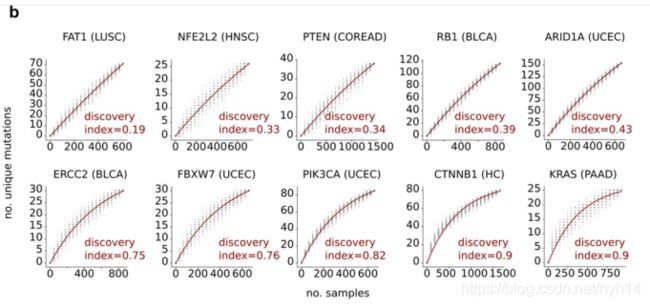
拿TP53为例,对于不同的癌症(不同颜色)的发现指数如图所示。
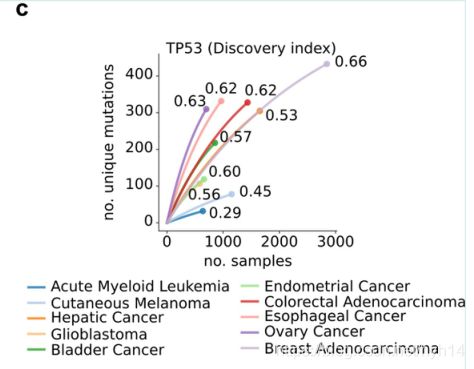
对于不同的基因名字(癌症名字)的100次迭代(重采样)发现指数的分布,取其中值。(对于一个基因(癌症),由于样本序列的不同,基因突变的发现指数会有不同,100次迭代产生100个发现指数,就会产生分布。)

对于TP53来说也是如此。

282个基因(癌症)构成训练数据的正集,相应的负集是通过在同一基因上的合成突变进行整合。我们根据癌症基因发生肿瘤机制的18个特征,为282个基因组织组合中的每一个建立了特定的模型(梯度增强树)。补充表1中包含18个特征介绍(Consequence type(4个)、Linear Clusters(3个)、3D clusters(2个)、Enriched protein domains(2个)、Phylogenetic conservation(1个)、Post-translational modifications(5个)、NMD skipping nonsense(1个)共18个)。 (结果类型、线性聚类、3D聚类、富集蛋白质域、系统保护、转录后修饰、NMD)
补充表1 https://static-content.springer.com/esm/art%3A10.1038%2Fs41586-021-03771-1/MediaObjects/41586_2021_3771_MOESM3_ESM.xlsx
术语:SNP Summary Columns

上图为肺腺癌(LUAD) EGFR boostDM模型的训练和交叉验证。
我们训练50个基分类器在相同数目的正突变和负突变的随机子集上,表示乘客突变的多样性和防止过拟合。集成到一个组合模型中(boostDM),该模型可以对组织中所有可能的癌症基因突变进行分类,并且给予解释。

上图为18个特征对EGFRL858R驱动突变分类的贡献。在放射状图中,在0线(内圆)上方出现代表有积极贡献的特征。
模型的性能
文章通过交叉验证来测试boostDM的性能,其中每个随机子集的测试突变由相应的基分类器分析。
补充说明:
为了衡量二分类器的分类性能,我们使用了加权F评分(F50),它更加重视精度(precision,P)而并不是召回率(recall,R)。
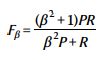 取β=0.5。
取β=0.5。
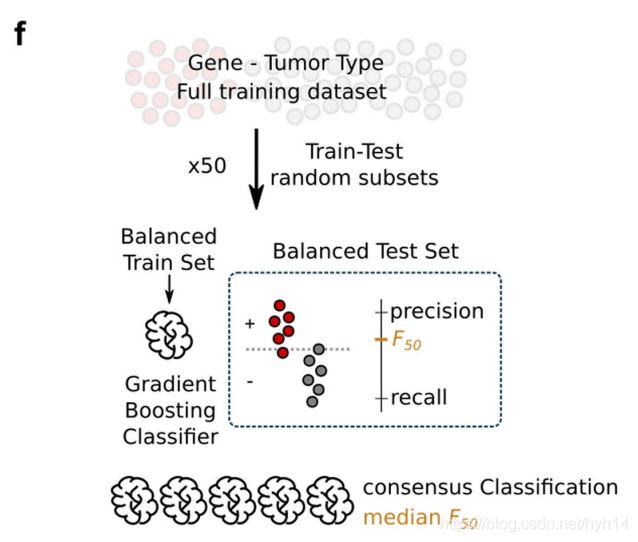
将观察到的突变和合成突变的50个子集随机分为训练集和测试机,并用于交叉验证,从中得到F50的一组值。

在50种癌症基因组合中观察到的突变比例(所有突变,包括同义突变),这些基因组合被相应的boostDM分类为驱动因子。令人欣慰的是,并没有把全部突变都归为驱动突变。
左:282个组合的F50与发现指数之间的关系,致癌基因(棕色)、肿瘤抑制基因(绿色)的分布。Q1区域(F50处于0.91和1之间,发现指数低于0.58.)右:特征复杂性指数的累计分布。特征复杂指数:分类器编码规则的复杂性(在补充说明)。
一般来说,由更大的训练集和更大的发现指数构建的模型表现出更好的性能。44个发现指数较低的基因组织组合模型(低于0.58;图1c中的Q1)的F50值大于0.91。这些模型大多数代表肿瘤抑制基因,在特征组成方面表现出较低的复杂性(图1c,扩展数据图2a),表明相对简单的特征组合能够准确地描述它们的驱动突变。


18个突变特征的SHAP值的可压缩性评估计算模型复杂性指数。曲线下的面积越大,复杂指数越小。
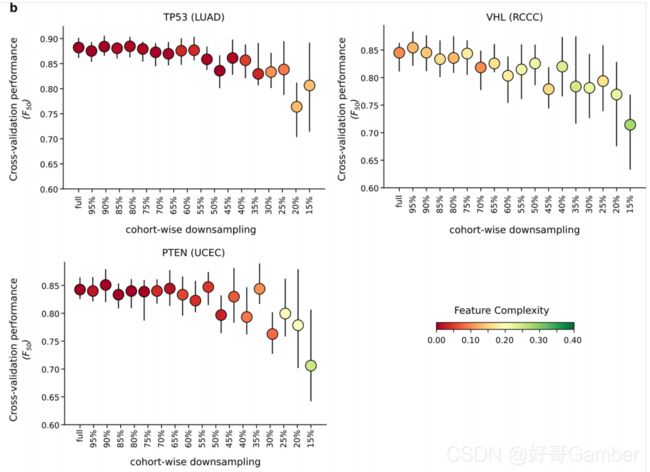
对可用于训练的肿瘤集进行子抽样显示,模型的性能随着训练集的大小而增加(扩展数据图2b),这表明,随着对更多的肿瘤队列进行排序,可用模型的数量和质量将增加。
为了进行跨肿瘤类型癌症基因的实验(across tumour types),我们选择了185个交叉验证F50值大于0.8且其驱动突变具有良好代表性的模型(扩展数据图1a,补充表2,补充说明)。
跨模型实验测试,在对从未在训练或交叉验证中见过的样本中的突变进行分类的现实场景中,模型的评估产生了与交叉验证中获得的相同或更高的F50值(扩展数据图2c(跨TP53模型))。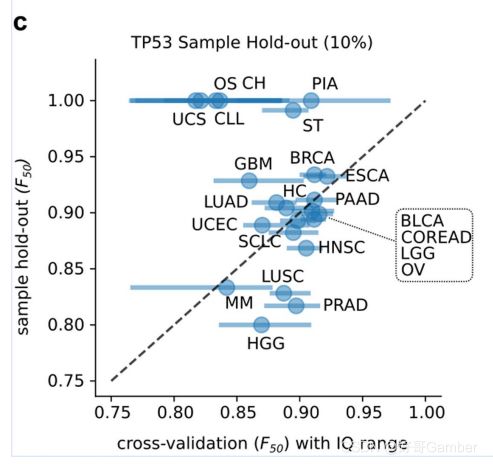
比较TP53的高信任度模型的交叉验证的性能(从基分类器获得的F50的中位数和四分位数)与随机模型训练和测试样本的90%每个肿瘤类型,而剩下的10%作为外部数据集进行验证。
这表明模型比基分类器具有更高的鉴别能力(扩展数据图2d)。
155个癌症基因肿瘤类型组合的交叉验证和再训练模型的中位数F50分布(如d),其中90%的原始样本再训练是可能的,符合扩展数据图1a所列的条件。箱线图:中线、中位数;箱限,第一和第三四分位数;须,第一四分位数的最低/最高数据点减/加1.5 IQR。
BoostDM模型显示了非常好的性能(F50>0.92),用于对经实验验证的癌基因中罕见突变的分类13,14(模型训练中排除;图1 d)。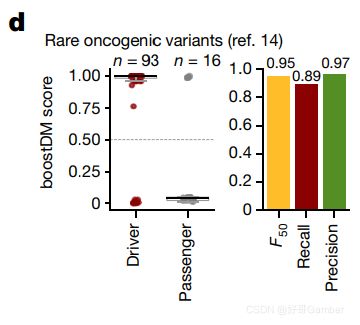
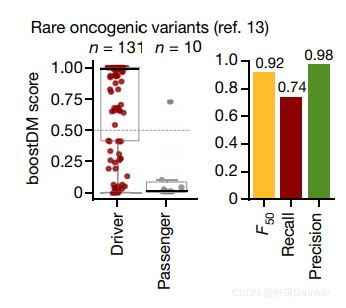
BoostDM评分(左)和绝对表现(右)用于分类通过两项实验分析确定的影响致癌基因的罕见致癌和良性变异。
我们还比较了TP53、KRAS、NRAS、HRAS和PTEN模型在肿瘤类型中的表现,以及四种不同的饱和诱变试验(11、12、15、16、22)。值得注意的是,在这个比较中,我们使用了基本分类器而不是模型,确保测试突变从来不是训练的一部分。所有的boostDM模型测试的结果优于饱和诱变试验。BoostDM模型的表现也优于7种旨在识别驱动器突变或评估其功能影响的计算方法。补充说明),并在肿瘤抑制基因的实验验证突变和注释的致病和良性变异的分类中表现良好(扩展数据图4a c)。

图内,罕见但经过实验验证的致癌和良性突变的boostDM评分和boostDM模型的性能(精度、召回率和F50)。
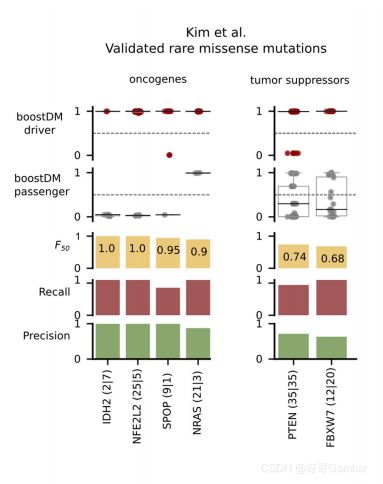
具体的性能。括号内是正集和负集的大小。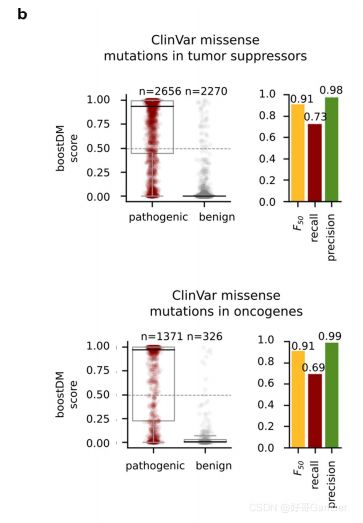
boostDM模型(精度、召回率、F50)在区分致病性体细胞突变和良性种系突变上的表现。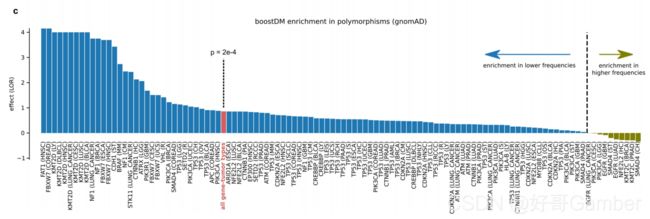
预测驱动因子在不同等位基因频率的多态性中的分布。条形图表示多态性的分类频率对其boostDM分类的逻辑回归效应大小。具有正效应大小的条形图代表基因(或汇集的癌症基因,红色,有回归P值表示),在这些基因中,非常罕见的多态性被boostDM模型分类为驱动基因的可能性增加。显示了与所有癌症基因的多态性相对应的逻辑回归的P值。
驱动突变的特征
观察到的癌症基因突变在两种癌症中可能不同,反映出不同癌症的发生机制。例如,图a中突变对于EGFR在LUAD和GBM上的突变分类很重要,但是这些突变会影响每个蛋白质的不同区域。图b中的Pkinase domain对于LUAD有很大的贡献,但是在GBM中没有。
在肺腺癌(LUAD,上)和胶质母细胞瘤(GBM,下)中,观察到的突变(驱动程序为红色圆圈)沿EGFR序列的分布。插图中,所有或唯一观察到的突变的部分被归类为两种恶性肿瘤的驱动因子(红色片段)。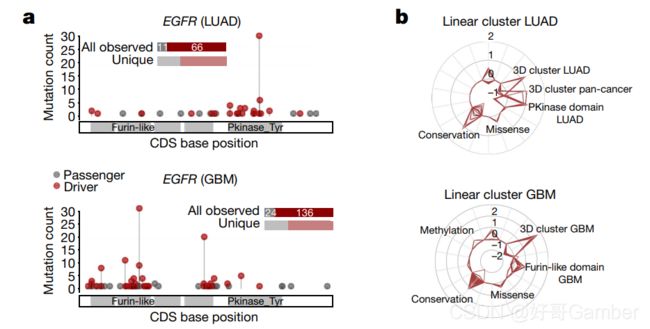

一些特征,如 mutational clusters用来识别重要的蛋白质区域,functionally important residues, or degrons 对许多突变的分类有重要贡献。
热图表示突变特征对185高置信度模型识别为驱动的所有突变的分类贡献。蓝色代表负SHAP (Shapley additive explanation)值(即对突变作为驱动因素的分类有负贡献的特征值),红色代表正值。突变被分成四组(上图)。LGG:脑低级别胶质瘤;LUSC:肺鳞状细胞癌;RCCC:肾透明细胞癌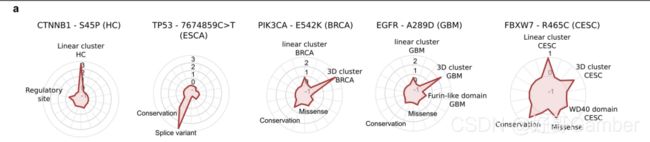
癌基因的驱动蓝图
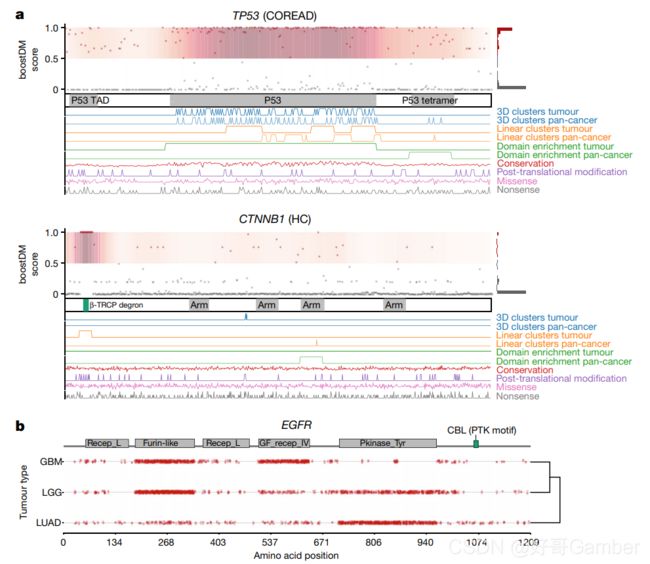
将185个基因癌症组合模型应用于in silico saturation mutagenesis中所有可能的(观察到的和未观察到的)核苷酸变化,得到了蛋白质不同区域在不同癌症中可能携带驱动突变的蓝图。解释为不同癌症的发生机制。BoostDM
结肠直肠癌中TP53所有可能突变的分类蓝图(COREAD;(上)和CTNNB1在肝癌(HC,下)中的表达。红色:潜在的驱动突变;灰色,旅客突变。每个蓝图下面突出显示了Pfam域的位置。在图的下方,彩色的轨迹代表了突变特征的分布,用来沿着蛋白质序列训练模型。直方图(右)显示了boostDM分数的分布。三种癌症的潜在驱动突变沿EGFR氨基酸序列的分布
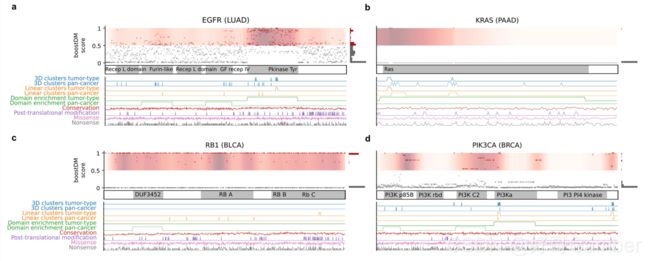
肺腺癌中EGFR (a)、胰腺腺癌中KRAS (b)、膀胱癌中RB1 (c)和乳腺腺癌中PIK3CA (d)的潜在驱动突变蓝图。所有癌症基因的模型蓝图可在https://intogen.org/ boostdm上获得。
比较肿瘤抑制基因和致癌基因中(来自所有可能的)潜在驱动突变的比例。前者比后者表现出更大比例的潜在驱动突变。

33种肿瘤类型中潜在驱动突变沿TP53序列的分布树形图代表了恶性肿瘤之间潜在驱动突变分布的相似性。
突变概率的影响
为了探讨突变概率对驱动突变的影响,基于在同一癌症中观察到的所有突变的三核苷酸频率分布,我们计算了所有在癌症中的潜在驱动突变的发生概率。突变概率偏差,观察到的突变和未观察到的突变的驱动突变概率分布纸巾的差异。概率偏差大于0.5表示观测突变的概率大于未观察到突变的概率。
突变概率---------的计算
背景知识
SNP位点:单核苷酸多态性位点,在一个能正常表达蛋白质的基因序列中,有些位置上的核苷酸不一定严格的是ACGT当中的一种,可以是两种、三种、或者全部。也就是说,把这个位置上的碱基替换成其他的,这个基因的功能还是正常的。这个位点叫SNP位点。
碱基突变类型
6种碱基组合与96种组合
碱基突变共有六类碱基取代:C-> A,C-> G,C-> T,T-> A,T-> C,T-> G。为什么只有6种呢?因为G> T取代被认为等同于C> A取代,因为不可能区分最初发生在哪条DNA链(正向或反向)上。因此,C> A和G> T替换都计为“ C> A”类的一部分。出于相同的原因,G> C,G> A,A> T,A> G和A> C突变被计为“ C> G”,“ C> T”,“ T> A”,“ T> C”和“ T> G”类。
从5'和3'相邻碱基(也称为侧翼碱基对或三核苷酸上下文)中获取信息会导致96种可能的突变类型(例如A [C> A] A,A [C> A] T等)。肿瘤的突变目录是通过将96种突变类型之一中的每个单核苷酸变体(SNV)分类(同义词:碱基对取代或置换点突变)并计算这96种突变类型中每种突变的总数来创建的。
对于每一个基因癌症组合,我们根据观察到的非驱动基因突变的突变谱(IntOGen)(序列形式)计算了每个位点上的每个三核苷酸上下文的突变率。
对于一个给定的基因癌症组合,每一个位点突变概率
其中,p(c)归一化后的频率,n(c)是基因编码序列中c环境的位点数,z贯穿所有96个以嘧啶为中心的三核苷酸上下文。那么选择的任意单位取决于基因的大小,但基因内的突变概率仍然是可比较的。