论文复现:DeepDTA: deep drug–target binding affinity prediction Hakime
论文复现,只实现模型及5-fold交叉验证,论文模型,未实现参数优化以及baseline对比。
- 数据读入:

- 模型:
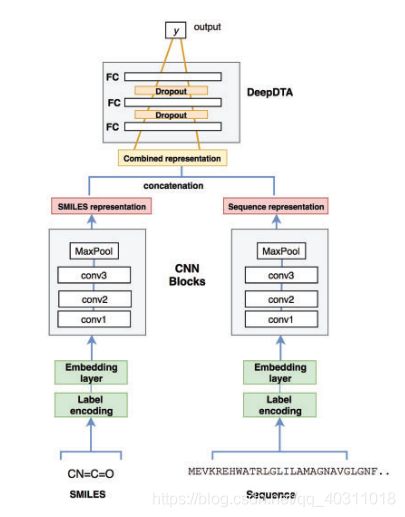
- 文件结构:

- datahelper.py 改自官方发布的代码
import numpy as np
import json
import pickle
import math
from collections import OrderedDict
# from keras.preprocessing.sequence import pad_sequences
## ######################## ##
#
# Define CHARSET, CHARLEN
#
## ######################## ##
# CHARPROTSET = { 'A': 0, 'C': 1, 'D': 2, 'E': 3, 'F': 4, 'G': 5, 'H': 6, \
# 'I': 7, 'K': 8, 'L': 9, 'M': 10, 'N': 11, 'P': 12, 'Q': 13, \
# 'R': 14, 'S': 15, 'T': 16, 'V': 17, 'W': 18, 'Y': 19, 'X': 20, \
# 'O': 20, 'U': 20,
# 'B': (2, 11),
# 'Z': (3, 13),
# 'J': (7, 9) }
# CHARPROTLEN = 21
CHARPROTSET = {"A": 1, "C": 2, "B": 3, "E": 4, "D": 5, "G": 6,
"F": 7, "I": 8, "H": 9, "K": 10, "M": 11, "L": 12,
"O": 13, "N": 14, "Q": 15, "P": 16, "S": 17, "R": 18,
"U": 19, "T": 20, "W": 21,
"V": 22, "Y": 23, "X": 24,
"Z": 25}
CHARPROTLEN = 25
CHARCANSMISET = {"#": 1, "%": 2, ")": 3, "(": 4, "+": 5, "-": 6,
".": 7, "1": 8, "0": 9, "3": 10, "2": 11, "5": 12,
"4": 13, "7": 14, "6": 15, "9": 16, "8": 17, "=": 18,
"A": 19, "C": 20, "B": 21, "E": 22, "D": 23, "G": 24,
"F": 25, "I": 26, "H": 27, "K": 28, "M": 29, "L": 30,
"O": 31, "N": 32, "P": 33, "S": 34, "R": 35, "U": 36,
"T": 37, "W": 38, "V": 39, "Y": 40, "[": 41, "Z": 42,
"]": 43, "_": 44, "a": 45, "c": 46, "b": 47, "e": 48,
"d": 49, "g": 50, "f": 51, "i": 52, "h": 53, "m": 54,
"l": 55, "o": 56, "n": 57, "s": 58, "r": 59, "u": 60,
"t": 61, "y": 62}
CHARCANSMILEN = 62
CHARISOSMISET = {"#": 29, "%": 30, ")": 31, "(": 1, "+": 32, "-": 33, "/": 34, ".": 2,
"1": 35, "0": 3, "3": 36, "2": 4, "5": 37, "4": 5, "7": 38, "6": 6,
"9": 39, "8": 7, "=": 40, "A": 41, "@": 8, "C": 42, "B": 9, "E": 43,
"D": 10, "G": 44, "F": 11, "I": 45, "H": 12, "K": 46, "M": 47, "L": 13,
"O": 48, "N": 14, "P": 15, "S": 49, "R": 16, "U": 50, "T": 17, "W": 51,
"V": 18, "Y": 52, "[": 53, "Z": 19, "]": 54, "\\": 20, "a": 55, "c": 56,
"b": 21, "e": 57, "d": 22, "g": 58, "f": 23, "i": 59, "h": 24, "m": 60,
"l": 25, "o": 61, "n": 26, "s": 62, "r": 27, "u": 63, "t": 28, "y": 64}
CHARISOSMILEN = 64
## ######################## ##
#
# Encoding Helpers
#
## ######################## ##
# Y = -(np.log10(Y/(math.pow(math.e,9))))
def one_hot_smiles(line, MAX_SMI_LEN, smi_ch_ind):
X = np.zeros((MAX_SMI_LEN, len(smi_ch_ind))) # +1
for i, ch in enumerate(line[:MAX_SMI_LEN]):
X[i, (smi_ch_ind[ch] - 1)] = 1
return X # .tolist()
def one_hot_sequence(line, MAX_SEQ_LEN, smi_ch_ind):
X = np.zeros((MAX_SEQ_LEN, len(smi_ch_ind)))
for i, ch in enumerate(line[:MAX_SEQ_LEN]):
X[i, (smi_ch_ind[ch]) - 1] = 1
return X # .tolist()
def label_smiles(line, MAX_SMI_LEN, smi_ch_ind):
X = np.zeros(MAX_SMI_LEN)
for i, ch in enumerate(line[:MAX_SMI_LEN]): # x, smi_ch_ind, y
X[i] = smi_ch_ind[ch]
return X # .tolist()
def label_sequence(line, MAX_SEQ_LEN, smi_ch_ind):
X = np.zeros(MAX_SEQ_LEN)
for i, ch in enumerate(line[:MAX_SEQ_LEN]):
X[i] = smi_ch_ind[ch]
return X # .tolist()
## ######################## ##
#
# DATASET Class
#
## ######################## ##
def parse_data(p, with_label=True):
fpath = p['fpath']
print("Read %s start" % fpath)
ligands = json.load(open(fpath + "ligands_can.txt"), object_pairs_hook=OrderedDict)
proteins = json.load(open(fpath + "proteins.txt"), object_pairs_hook=OrderedDict)
Y = pickle.load(open(fpath + "Y", "rb"), encoding='latin1') ### TODO: read from raw
is_log = p['is_log']
if is_log:
Y = -(np.log10(Y / (math.pow(10, 9))))
XD = []
XT = []
if with_label:
for d in ligands.keys():
XD.append(label_smiles(ligands[d], p['smilen'], CHARISOSMISET))
for t in proteins.keys():
XT.append(label_sequence(proteins[t], p['seqlen'], CHARPROTSET))
else:
for d in ligands.keys():
XD.append(one_hot_smiles(ligands[d], p['smilen'], CHARISOSMISET))
for t in proteins.keys():
XT.append(one_hot_sequence(proteins[t], p['seqlen'], CHARPROTSET))
return XD, XT, Y
- train.py , 论文提供的有两个数据集,此次使用的KIBA数据集,5-fold交叉验证结果未取平均和test(懒~)
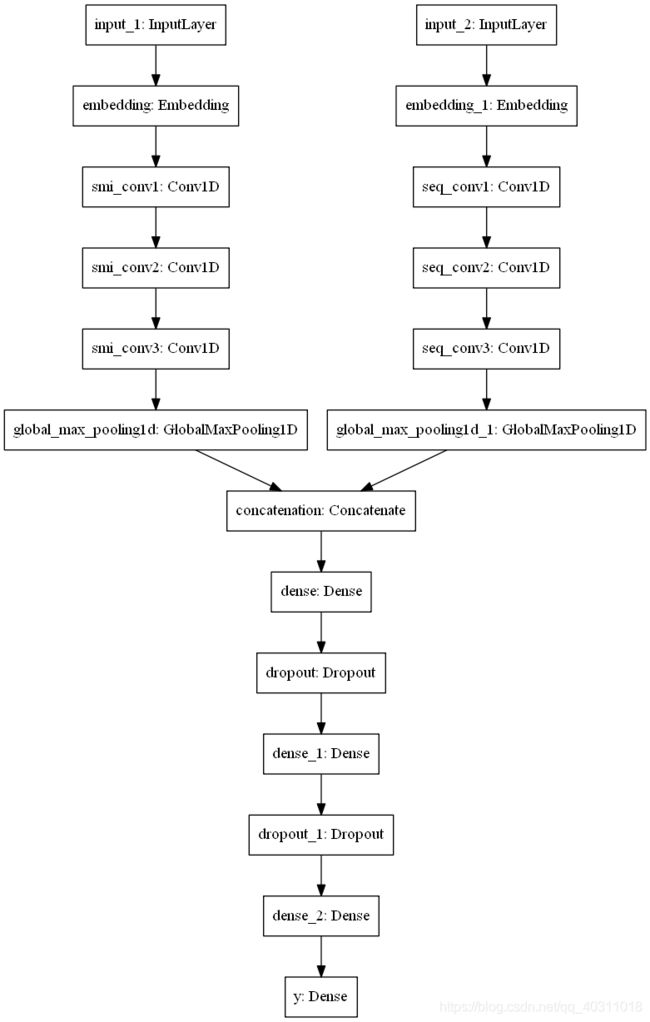
import tensorflow as tf
import numpy as np
import pickle
import json
import copy
import math
import datahelper as dhlper
# label filePath of kiba
kiba_label_path = './data/kiba/Y'
davis_label_path = './data/davis/Y'
# train and test data filePath of kiba
kiba_train_path = './data/kiba/folds/train_fold_setting1.txt'
kiba_test_path = './data/kiba/folds/test_fold_setting1.txt'
def CNNs_DeepDTA_Net(max_seq_len, max_smi_len, num_filters, filter_length1, filter_length2):
# Input shape
d_input = tf.keras.Input(shape=(max_smi_len,), dtype='int32')
p_input = tf.keras.Input(shape=(max_seq_len,), dtype='int32')
# CNN block of drug representation
d_p = tf.keras.layers.Embedding(input_dim=dhlper.CHARISOSMILEN + 1, output_dim=128,
input_length=max_smi_len)(d_input)
d_p = tf.keras.layers.Conv1D(filters=num_filters, kernel_size=filter_length1, activation='relu', padding='valid',
strides=1, name='smi_conv1')(d_p)
d_p = tf.keras.layers.Conv1D(filters=num_filters * 2, kernel_size=filter_length1, activation='relu',
padding='valid', strides=1, name='smi_conv2')(d_p)
d_p = tf.keras.layers.Conv1D(filters=num_filters * 3, kernel_size=filter_length1, activation='relu',
padding='valid', strides=1, name='smi_conv3')(d_p)
d_p = tf.keras.layers.GlobalMaxPool1D()(d_p)
# CNN block of protein representation
p_p = tf.keras.layers.Embedding(input_dim=dhlper.CHARPROTLEN + 1, output_dim=128,
input_length=max_seq_len)(p_input)
p_p = tf.keras.layers.Conv1D(filters=num_filters, kernel_size=filter_length2, activation='relu', padding='valid',
strides=1, name='seq_conv1')(p_p)
p_p = tf.keras.layers.Conv1D(filters=num_filters * 2, kernel_size=filter_length2, activation='relu',
padding='valid', strides=1, name='seq_conv2')(p_p)
p_p = tf.keras.layers.Conv1D(filters=num_filters * 3, kernel_size=filter_length2, activation='relu',
padding='valid', strides=1, name='seq_conv3')(p_p)
p_p = tf.keras.layers.GlobalMaxPool1D()(p_p)
# Combined representation
c_p = tf.keras.layers.concatenate([d_p, p_p], axis=-1, name='concatenation')
# three layers of full connection
FC = tf.keras.layers.Dense(1024, activation='relu')(c_p)
FC = tf.keras.layers.Dropout(0.1)(FC)
FC = tf.keras.layers.Dense(1024, activation='relu')(FC)
FC = tf.keras.layers.Dropout(0.1)(FC)
FC = tf.keras.layers.Dense(512, activation='relu')(FC)
y = tf.keras.layers.Dense(1, kernel_initializer='normal', name='y')(FC)
model = tf.keras.Model(inputs=[d_input, p_input], outputs=[y])
model.compile(optimizer='adam', loss='mean_squared_error', metrics=[cindex_score])
print(model.summary())
tf.keras.utils.plot_model(model, to_file='./result/network.png')
return model
# from paper
def cindex_score(y_true, y_pred):
g = tf.subtract(tf.expand_dims(y_pred, -1), y_pred)
g = tf.cast(g == 0.0, tf.float32) * 0.5 + tf.cast(g > 0.0, tf.float32)
f = tf.subtract(tf.expand_dims(y_true, -1), y_true) > 0.0
f = tf.linalg.band_part(tf.cast(f, tf.float32), -1, 0)
g = tf.reduce_sum(tf.multiply(g, f))
f = tf.reduce_sum(f)
return tf.where(tf.equal(g, 0), 0.0, g / f) # select
# from paper
def get_cindex(Y, P):
summ = 0
pair = 0
for i in range(1, len(Y)):
for j in range(0, i):
if i is not j:
if Y[i] > Y[j]:
pair += 1
summ += 1 * (P[i] > P[j]) + 0.5 * (p[i] == p[j])
if pair is not 0:
return summ / pair
else:
return 0
# from paper
def get_pairs_binding_affinity(XD, XT, Y, rows, cols):
drugs = []
targets = []
affinity = []
for pair_ind in range(len(rows)):
drug = XD[rows[pair_ind]]
drugs.append(drug)
target = XT[cols[pair_ind]]
targets.append(target)
affinity.append(Y[rows[pair_ind], cols[pair_ind]])
drug_data = np.stack(drugs)
target_data = np.stack(targets)
return drug_data, target_data, affinity
def experiment(train_path, test_path, flodcount=5): # 5-fold cross validation and test
p = {'fpath': './data/kiba/', 'setting_no': 1, 'seqlen': 1000, 'smilen': 100,
'is_log': False}
XD, XT, Y = dhlper.parse_data(p)
label_drug_inds, label_protein_inds = np.where(np.isnan(Y) == False)
# np.where(condition, x, y) If the condition is satisfied, output x, else output y
# label_drug_inds --drug index
# label_protein_inds --protein index
test_fold = json.load(open(test_path))
train_folds = json.load(open(train_path)) # ----
# test dataset, 1份
# test_drug_indices = label_drug_inds[test_fold]
# test_protein_indices = label_protein_inds[test_fold]
# train datasets(contain five lists), 5
# train_drug_indices = label_drug_inds[train_folds[2]]
# train_protein_indices = label_protein_inds[train_folds[2]]
# max_smi_len drug
max_smi_len = 100
# max_seq_len protein
max_seq_len = 1000
# num_filters
num_filters = 32
# filter_lenghth1 (drug CNN block)
filter_length1 = 4
# filter_lenghth2 (protein CNN block)
filter_length2 = 8
# some arguments
batch_size = 256
epochs = 100
test_sets = []
train_sets = []
val_sets = []
# 5-fold cross valuation data
for i in range(flodcount):
val_flod = train_folds[i]
val_sets.append(val_flod)
copytrain = copy.deepcopy(train_folds)
copytrain.pop(i)
otherfoldsinds = [item for sublist in copytrain for item in sublist]
train_sets.append(otherfoldsinds)
test_sets.append(test_fold)
# print("val set", str(len(val_flod)))
# print("train set", str(len(otherfoldsinds)))
for flodIndex in range(len(val_sets)):
valIndex = val_sets[flodIndex]
trainIndex = train_sets[flodIndex]
trrows = label_drug_inds[trainIndex]
trcols = label_protein_inds[trainIndex]
train_drugs, train_proteins, train_Y = get_pairs_binding_affinity(XD, XT, Y, trrows, trcols)
print(np.array(train_drugs).shape)
print(np.array(train_proteins).shape)
terows = label_drug_inds[valIndex]
trcols = label_protein_inds[valIndex]
val_drugs, val_proteins, val_Y = get_pairs_binding_affinity(XD, XT, Y, terows, trcols)
# loading model
model = CNNs_DeepDTA_Net(max_seq_len, max_smi_len, num_filters, filter_length1, filter_length2)
es = tf.keras.callbacks.EarlyStopping(monitor='val_loss', mode='min', patience=10, verbose=1)
gridres = model.fit(x=([np.array(train_drugs), np.array(train_proteins)]), y=np.array(train_Y),
batch_size=batch_size, epochs=epochs,
validation_data=([np.array(val_drugs), np.array(val_proteins)], np.array(val_Y)),
shuffle=False, callbacks=[es])
predicted_labels = model.predict([np.array(val_drugs), np.array(val_proteins)])
loss, CIndex_ii = model.evalute(x=([np.array(val_drugs), np.array(val_proteins)]), y=np.array(val_Y), verbose=0)
CIndex_i = get_cindex(val_Y, predicted_labels)
print('Fold = %d, CI_i = %.4f, CI_ii = %.4f, MSE = %.4f' % (flodIndex, CIndex_i, CIndex_ii, loss))
# the average…………
# test………………
experiment(kiba_train_path, kiba_test_path)
- 结果: emmm~对比论文结果,还行
