- 学点心理知识,呵护孩子健康
静候花开_7090
昨天听了华中师范大学教育管理学系副教授张玲老师的《哪里才是学生心理健康的最后庇护所,超越教育与技术的思考》的讲座。今天又重新学习了一遍,收获匪浅。张玲博士也注意到了当今社会上的孩子由于心理问题导致的自残、自杀及伤害他人等恶性事件。她向我们普及了一个重要的命题,她说心理健康的一些基本命题,我们与我们通常的一些教育命题是不同的,她还举了几个例子,让我们明白我们原来以为的健康并非心理学上的健康。比如如果
- 【一起学Rust | 设计模式】习惯语法——使用借用类型作为参数、格式化拼接字符串、构造函数
广龙宇
一起学Rust#Rust设计模式rust设计模式开发语言
提示:文章写完后,目录可以自动生成,如何生成可参考右边的帮助文档文章目录前言一、使用借用类型作为参数二、格式化拼接字符串三、使用构造函数总结前言Rust不是传统的面向对象编程语言,它的所有特性,使其独一无二。因此,学习特定于Rust的设计模式是必要的。本系列文章为作者学习《Rust设计模式》的学习笔记以及自己的见解。因此,本系列文章的结构也与此书的结构相同(后续可能会调成结构),基本上分为三个部分
- 四章-32-点要素的聚合
彩云飘过
本文基于腾讯课堂老胡的课《跟我学Openlayers--基础实例详解》做的学习笔记,使用的openlayers5.3.xapi。源码见1032.html,对应的官网示例https://openlayers.org/en/latest/examples/cluster.htmlhttps://openlayers.org/en/latest/examples/earthquake-clusters.
- 特殊的拜年
飘雪的天堂
文/雪儿大年初一,家家户户没有了轰响的鞭炮声,大街上没有了人流涌动的喧闹,几乎看不到人影,变得冷冷清清。天刚亮不大会儿,村里的大喇叭响了起来:由于当前正值疾病高发期,流感流行的高峰期。同时,新型冠状病毒感染的肺炎进入第二波流行的上升期。为了自己和他人的健康安全着想,请大家尽量不要串门拜年,不要在街里走动。可以通过手机微信,视频,电话,信息拜年……今年的春节真是特别。禁止燃放鞭炮,烟花爆竹,禁止出村
- CX8903:Ebike自行车仪表电源方案开发,Ebike智能仪表电源芯片
诚芯微科技
社交电子
CX8903:电动Ebike自行车仪表电源方案开发,Ebike智能仪表电源芯片推荐。电动助力自行车EBIKE凭借其环保、健康、低噪、和便捷等特点,成为了越来越受欢迎的骑行便利交通工具。提供电动Ebike自行车仪表电源方案开发、E-BIKE电动助力自行车仪表供电电源解决方案。CX8903采用100V高压制造工艺(芯片最高耐压可到100V以上),SOP-8L贴片封装,CX8903内置100V/90mΩ
- 18、架构-可观测性之聚合度量
大树~~
架构javapython后端架构
聚合度量聚合度量是指对系统运行时产生的各种指标数据进行收集、聚合和分析,以了解系统的健康状况和性能表现。聚合度量是可观测性的关键组成部分,通过对度量数据的分析,可以及时发现系统中的异常和瓶颈。以下是对聚合度量各个方面的详细解析,并结合具体的数据案例和技术支撑。指标收集收集系统运行时产生的各种指标数据是聚合度量的基础。常见的指标包括CPU使用率、内存使用率、请求处理时间、请求数、错误率等。以下是指标
- 王东伟,中原焦点秦皇岛站第五期,每日分享第181天
Vivian_c8c7
《解码青春期》让孩子懂得承担责任,学会道歉。英国诗人亚历山大•蒲柏有句名言:凡人难免犯错宽恕方显神性。学会如何请求对方宽恕对于保持健康的关系至关重要。当青少年把事情搞砸的时候,他们需要从关心他们的成年人那里获得帮助。家长的目标是要培养一个能为自己的行为承担责任的青少年,培养一个敢于诚恳的承认错误,愿意真心悔改的青少年。青少年只关注自己如何委屈,而且会竭尽全力为自己的行为辩解。所以,家长得小心地拆除
- 社保应该缴15年还是25年?那种方式最划算?
袋鼠观保保险规划师
社保无论是缴费15年还是25年,影响最大的就是养老保险和医疗保险,缴费时间越长越有利!1.养老保险真的交满15年就够了吗?要知道,社保缴费时长,直接影响到退休后能拿多少养老金,而且交得越久,退休领得越多。我拿深圳作为例子,想拿到养老金必须满足两个条件:只要达到一定的退休年龄,养老保险累计交满15年就可以拿到养老金了。那如果多缴了20年、25年甚至30年,是不是浪费了?实际上,缴满15年只是刚好可以
- “这才好”麻辣香锅 能够增加人身体的免疫能力
小补文知
我就来介绍一种香锅,那就是“这才好”麻辣香锅,它产出于著名的蜀地文化,具有悠久的历史土家风味,麻辣鲜香,健康安全。采用传统秘制麻辣香锅油辣子,还有贴心加料“孜然包”满足人们的不同口味需求,香锅底料辣椒,微辣且香,含有丰富微量元素和维生素,具有辣而不躁,味道纯正,醇厚温和。花椒采用历史悠久,被列为宫廷供品的“贡椒”的汉源花椒。我们还挑选了“川菜之魂”郫县豆瓣的鼻祖品牌豆瓣,保留最原始的郫县豆瓣味道,
- 《度五行》生活报报甲午62:不通痛苦,太通也痛苦,要健康快乐,需要通体舒畅。
YangduSam2021
220809壬寅戊申甲午,《度.生活五行》:天干土克水,水生木,木克土。地支寅申冲,寅午合。20220809,周二,兴大上海六班2512天,西交大2013上海班3212天,后TA15332天,度生活619天,今天拜访了一家有趣且当红产业的新创公司AK。AK一开始从事深海新能源储存与供电设备的研发生产制造,2年前开始做移动与家庭储能设备的研发生产制造。觉得有趣是因为这是笔者认知里用科技做降维打击的公
- 父母教育孩子的方式,将影响孩子一生
树英教育
为什么有些孩子总是充满自信与快乐?独立、有主见又坚强?而有些孩子却自卑、胆怯,软弱又过度依赖父母?为什么有些孩子总是健康、阳光又富于创造力?而有些孩子却悲观、孤僻又思想空乏?一个孩子的行为取决于孩子的思想,思想取决于环境和自己的认知,认知取决于教育。父母是孩子人生中的第一位教育者,父母养育孩子的方式,将决定他们人生的高度,影响他们的一生。网络图,侵权即删优秀的父母就像园丁,既要浇水施肥,又要修剪杂
- 2024.9.6 Python,华为笔试题总结,字符串格式化,字符串操作,广度优先搜索解决公司组织绩效互评问题,无向图
RaidenQ
python华为leetcode算法力扣广度优先无向图
1.字符串格式化name="Alice"age=30formatted_string="Name:{},Age:{}".format(name,age)print(formatted_string)或者name="Alice"age=30formatted_string=f"Name:{name},Age:{age}"print(formatted_string)2.网络健康检查第一行有两个整数m
- 2020-8-19晨间日记:看过的电影
盐大虾
今天是周三起床:6点半就寝:11点天气:晴心情:正常纪念日:周三任务清单今日完成的任务,最重要的三件事:1.整理写过的文档2.电影《电灯泡》3.这就是街舞第三季第五期改进:早睡早起习惯养成:早睡早起,看书周目标·完成进度两篇文章学习·信息·阅读电影艺术发展史相关教材健康·饮食·锻炼吃了挺多零食,还喝了果粒橙,还是得少吃,多锻炼,不然会慢慢死掉的。人际·家人·朋友淡定交流,不放在心上。工作·思考专心
- 快乐春节
Lilywo
春节肯定是大多小朋友都喜欢的节日吧,因为春节的娱乐项目可多啦,下面我就带大家去看一看某些娱乐项目吧!第一件肯定就是穿新衣啦!因为辞旧迎新,一年过去了,要迎来新的一年。所以过年穿新衣也是一项习俗吧;第二件,收压岁钱。压岁钱大家都知道吧,过年的时候,小朋友们肯定都会受到大人们的压岁钱吧,对啦!大家知道为什么亲人们会给我们压岁钱呢?答案是因为亲人们希望我们在新的一年里可以健健康康、平平安安,幸福福的生活
- 语文主题教学学习笔记之87
东哥杂谈
“语文主题教学”学习笔记之八十七(0125)今天继续学习小学语文主题教学的实践样态。板块三:教学中体现“书艺”味道。作为四大名著之一的《水浒传》,堪称我国文学宝库之经典。对从《水浒传》中摘选的单元,教师就要了解其原生态,即评书体特点。这也要求教师要了解一些常用的评书行话术语,然后在教学时适时地加入一些,让学生体味其文本中原有的特色。学生也要尽可能地通过朗读的方式,而不单是分析讲解的方式进行学习。细
- 2019-03-22
430O70Mk
引发支原体的原因支原体感染是临床上比较常见的一种疾病,此疾病会对患者的身体造成很大的伤害,对支原体感染患者的日常生活也会带来极大的影响,那么诱发支原体感染的原因是什么呢?肺炎支原体感染,又称支原体性肺炎,是由肺炎支原体引起的急性间质性肺炎。主要通过呼吸道传播,健康人吸入患者咳嗽,打喷嚏时喷出的口、鼻分泌物而感染。支原体为动物多种疾病的致病体,而其中只有肺炎支原体肯定对人致病。它是由口、鼻分泌物经空
- “愿你,戒酒也戒她。”
钟斯
你以荨麻疹为幌子,向全世界宣布,从今以后你不能再喝酒了,你要戒酒。你的朋友们,觉得你的健康最重要,所以大力支持,商量着以后谁都不能再劝你酒。可是,你转身就在夜深人静时独自痛饮,然后在意识涣散中发了疯地想她。你知道这样不对,但你难以自制。我一直觉得,你是那种“衣履风流,潇洒潇洒去了”的人,所以,当你跟我说你难受的时候,我还是有一些诧异的。我不知道,什么样的事情能让如此洒脱的你难受。当我问及的时候,你
- 到现在才发现自己有病
骑着大脑去南极
原来人们真的喜欢对某一概念设限的,为什么这么说的,如果说工作,你可能在脑子浮现出的工作的大概印象就是被动的干着不喜欢的活,被动拿着那些微薄的工资的场景。但是也不排除可能把工作想成成长的样子的人,只是这种物种很少吧。就比如像病这个概念,大部分可能仅仅就把他局限在身体上出现的异常不舒服状况称之为病,直到近代才把病拓展到精神层面。。。最近才发现原来在精神层面上是有病的,原来曾经认为自己是一个无比健康的小
- 3286、穿越网格图的安全路径
Lenyiin
题解c++算法leetcode
3286、[中等]穿越网格图的安全路径1、题目描述给你一个mxn的二进制矩形grid和一个整数health表示你的健康值。你开始于矩形的左上角(0,0),你的目标是矩形的右下角(m-1,n-1)。你可以在矩形中往上下左右相邻格子移动,但前提是你的健康值始终是正数。对于格子(i,j),如果grid[i][j]=1,那么这个格子视为不安全的,会使你的健康值减少1。如果你可以到达最终的格子,请你返回tr
- 高考后该不该给孩子买电脑,什么情况能买?什么情况不能买?
寻求改变
我知道家长们很担心,怕买了电脑小孩沉迷游戏,耽误了学业,也不利于身体健康。对于准大学生来说,基本上在18岁左右,也不算小了,但在很多父母眼里,依旧是个小孩子。数据显示,这种情况是有发生的,大学生约70%的电脑主要被用于玩网络游戏,如果没有养成一个用良好的习惯,对孩子影响是非常大的。我总结为三买,三不买。最近有看到群里很多家长再问,小孩上大学该不该给他买电脑,要买和不买两种观点的家长都有,那么哪种情
- 感恩日记7.24
初九强大记
感恩爸妈生我养我给我生命,用爱抚养我长大,让我有一个健康的身体、美丽的容颜、聪明的大脑。感恩爸妈现在身体依然健康,让我可以有更多的时间做自己想做的事,赚更多的钱。赞自己昨天去办公室以后,给大家拖地、打水,种下爱护生命的种子,回向给我的目标有一个健康快乐的小包宝,帮助大家节约了时间,有一个干净的空间,回向给我的目标2021年9月,工作转正,新工作离家近,时间自由。赞自己昨天给老公做好早餐,种下了爱护
- 放松的一天
4da9b7687fa0
20190325总结起床07:20图片发自App睡觉:23:00天气:晴今日任务清单学习·信息·阅读•水滴阅读Day40Alice’sAdventuresinWonderlandChapter6.2图片发自App•BBC跟读训练营Day24图片发自App图片发自App图片发自App•潘多拉口语训练营Day6Wow.Whatabigboy!•文化知识学习今日无•阅读时间地狱健康·饮食·锻炼•饮食目标
- 《转介绍方法论》学习笔记
小可乐的妈妈
一、高效转介绍的流程:价值观---执行----方案一)转介绍发生的背景:1、对象:谁向谁转介绍?全员营销,人人参与。①员工的激励政策、客户的转介绍诱因制作客户画像:a信任;支付能力;意愿度;便利度(根据家长具备四个特征的个数分为四类)B性格分类C职业分类D年龄性别②执行:套路,策略,方法,流程2、诱因:为什么要转介绍?认同信任;多方共赢;传递美好;零风险承诺打动人心,超越期待。选择做教育,就是选择
- 2019-04-10
shuaigefeng
姓名:王林锋企业名称:三亚蔚蓝时代实业有限公司组别:420期努力6组【日精进打卡251天】【知~学习、诵读】《六项精进》2遍,累计256遍《大学》2遍,累计220遍【经典分享】1、想过成功、想过失败、也想过放弃。【行~实践】一、修身:(对自己个人)1.拍打腿部两侧50下,舌顶上颚50下。2.坚持诵读、阅读。3.坚持锻炼、按时睡觉起床。4.控制健康饮食,饭后走动30分钟。5.每天反省自己的思想和行为
- JAVA学习笔记之23种设计模式学习
victorfreedom
Java技术设计模式androidjava常用设计模式
博主最近买了《设计模式》这本书来学习,无奈这本书是以C++语言为基础进行说明,整个学习流程下来效率不是很高,虽然有的设计模式通俗易懂,但感觉还是没有充分的掌握了所有的设计模式。于是博主百度了一番,发现有大神写过了这方面的问题,于是博主迅速拿来学习。一、设计模式的分类总体来说设计模式分为三大类:创建型模式,共五种:工厂方法模式、抽象工厂模式、单例模式、建造者模式、原型模式。结构型模式,共七种:适配器
- 2023-4-6晨间日记
百里清风柏年醉
今天是什么日子起床:7:00就寝:10:30天气:阳光明媚心情:沉闷,忧心忡忡纪念日:无任务清单昨日完成的任务,最重要的三件事:看咨询工程师的书锻炼身体记75个单词改进:自己做饭习惯养成:看纸质书籍不刷抖音每天日更周目标·完成进度学习·信息·阅读健康·饮食·锻炼人际·家人·朋友保持与朋友交流,多认识、结交新的朋友工作·思考怎么做好向上管理该学习什么新的技能怎么与同事更好相处,更好地开展工作最美好的
- 6.0 践行打卡 D47
星月格格
去努力改变1.运动步行13000+8分钟腿部拉伸2.阅读《墨菲定律》第三章第三节:霍桑效应~适度发泄,才能轻装上阵“霍桑效应”这一概念,源自于1924年一个1933年间以哈佛大学心理专家乔治·埃尔顿·梅奥教授为首进行的一系列工厂工人的谈话实验研究。“霍桑效应”告诉我们,在工作,生活中总会产生数不清的情绪反应,其中很大一部分是负面的负面情绪的积累会影响人的精神和心情,不仅仅会影响个人健康,还会破坏人
- (缓解抑郁症状)中原焦点团队杨小杰坚持分享第226天2021-4-1
yxjlady
缓解抑郁症状1、不要总待在室内,抑郁严重的人,通常都不想出门2、抑郁性都有诱因或一个导火索,人不能战胜所有东西,要有取舍3、社交,抑郁症的人总是自己脑中不断的自言自语,自我否定等,出去社交就被迫被别的东西点拨了,深度抑郁没法走出自己的世界,思维走不出自己的怪圈4、锻炼让自己轻微出汗最佳,身心是一体时,身体有活力,精神很难不健康5、冥想冥想和社交一样,可以改变你的神经可塑性,一个沉溺在自己世界里的抑
- 新能源汽车 BMS 学习笔记篇—BMS 基本定义及分类
WPG大大通
其他笔记汽车BMS经验分享新能源电池
一、BMS定义1、概念:BMS(BatteryManagementSystem)即电池管理系统,其管理对象是二次电池(充电电池或蓄电池),其主要目的是电池的利用率,防止电池出现过度充电和过度放电,可应用于电动汽车、电瓶车、机器人、无人机等图片来源:腾讯网https://new.qq.com《标准普尔警告,电动汽车电池生产面临供应链和地缘政治风险》2、四大功能①感知和测量:检测电池的电压、电流、温度
- 2022-5-23《儿童纪律教育》培训
手捧鲜花_54e3
张子博春蕾八幼缺乏技能导致的问题,需要老师和家长教授儿童所需要的锻炼的技能。比如教授儿童如何处理情绪、与人相处以及有效的交流技巧。未满足的情感需要,如信任、尊重、爱与权利的需要,都应该让儿童充分得到满足时,才能解决问题。家庭互动与复杂的原因,需要教师建立以家庭为中心的实践,和家庭沟通,建立和谐的关系,为孩子的健康成长共同努力。
- scala的option和some
矮蛋蛋
编程scala
原文地址:
http://blog.sina.com.cn/s/blog_68af3f090100qkt8.html
对于学习 Scala 的 Java™ 开发人员来说,对象是一个比较自然、简单的入口点。在 本系列 前几期文章中,我介绍了 Scala 中一些面向对象的编程方法,这些方法实际上与 Java 编程的区别不是很大。我还向您展示了 Scala 如何重新应用传统的面向对象概念,找到其缺点
- NullPointerException
Cb123456
androidBaseAdapter
java.lang.NullPointerException: Attempt to invoke virtual method 'int android.view.View.getImportantForAccessibility()' on a null object reference
出现以上异常.然后就在baidu上
- PHP使用文件和目录
天子之骄
php文件和目录读取和写入php验证文件php锁定文件
PHP使用文件和目录
1.使用include()包含文件
(1):使用include()从一个被包含文档返回一个值
(2):在控制结构中使用include()
include_once()函数需要一个包含文件的路径,此外,第一次调用它的情况和include()一样,如果在脚本执行中再次对同一个文件调用,那么这个文件不会再次包含。
在php.ini文件中设置
- SQL SELECT DISTINCT 语句
何必如此
sql
SELECT DISTINCT 语句用于返回唯一不同的值。
SQL SELECT DISTINCT 语句
在表中,一个列可能会包含多个重复值,有时您也许希望仅仅列出不同(distinct)的值。
DISTINCT 关键词用于返回唯一不同的值。
SQL SELECT DISTINCT 语法
SELECT DISTINCT column_name,column_name
F
- java冒泡排序
3213213333332132
java冒泡排序
package com.algorithm;
/**
* @Description 冒泡
* @author FuJianyong
* 2015-1-22上午09:58:39
*/
public class MaoPao {
public static void main(String[] args) {
int[] mao = {17,50,26,18,9,10
- struts2.18 +json,struts2-json-plugin-2.1.8.1.jar配置及问题!
7454103
DAOspringAjaxjsonqq
struts2.18 出来有段时间了! (貌似是 稳定版)
闲时研究下下! 貌似 sruts2 搭配 json 做 ajax 很吃香!
实践了下下! 不当之处请绕过! 呵呵
网上一大堆 struts2+json 不过大多的json 插件 都是 jsonplugin.34.jar
strut
- struts2 数据标签说明
darkranger
jspbeanstrutsservletScheme
数据标签主要用于提供各种数据访问相关的功能,包括显示一个Action里的属性,以及生成国际化输出等功能
数据标签主要包括:
action :该标签用于在JSP页面中直接调用一个Action,通过指定executeResult参数,还可将该Action的处理结果包含到本页面来。
bean :该标签用于创建一个javabean实例。如果指定了id属性,则可以将创建的javabean实例放入Sta
- 链表.简单的链表节点构建
aijuans
编程技巧
/*编程环境WIN-TC*/ #include "stdio.h" #include "conio.h"
#define NODE(name, key_word, help) \ Node name[1]={{NULL, NULL, NULL, key_word, help}}
typedef struct node { &nbs
- tomcat下jndi的三种配置方式
avords
tomcat
jndi(Java Naming and Directory Interface,Java命名和目录接口)是一组在Java应用中访问命名和目录服务的API。命名服务将名称和对象联系起来,使得我们可以用名称
访问对象。目录服务是一种命名服务,在这种服务里,对象不但有名称,还有属性。
tomcat配置
- 关于敏捷的一些想法
houxinyou
敏捷
从网上看到这样一句话:“敏捷开发的最重要目标就是:满足用户多变的需求,说白了就是最大程度的让客户满意。”
感觉表达的不太清楚。
感觉容易被人误解的地方主要在“用户多变的需求”上。
第一种多变,实际上就是没有从根本上了解了用户的需求。用户的需求实际是稳定的,只是比较多,也比较混乱,用户一般只能了解自己的那一小部分,所以没有用户能清楚的表达出整体需求。而由于各种条件的,用户表达自己那一部分时也有
- 富养还是穷养,决定孩子的一生
bijian1013
教育人生
是什么决定孩子未来物质能否丰盛?为什么说寒门很难出贵子,三代才能出贵族?真的是父母必须有钱,才能大概率保证孩子未来富有吗?-----作者:@李雪爱与自由
事实并非由物质决定,而是由心灵决定。一朋友富有而且修养气质很好,兄弟姐妹也都如此。她的童年时代,物质上大家都很贫乏,但妈妈总是保持生活中的美感,时不时给孩子们带回一些美好小玩意,从来不对孩子传递生活艰辛、金钱来之不易、要懂得珍惜
- oracle 日期时间格式转化
征客丶
oracle
oracle 系统时间有 SYSDATE 与 SYSTIMESTAMP;
SYSDATE:不支持毫秒,取的是系统时间;
SYSTIMESTAMP:支持毫秒,日期,时间是给时区转换的,秒和毫秒是取的系统的。
日期转字符窜:
一、不取毫秒:
TO_CHAR(SYSDATE, 'YYYY-MM-DD HH24:MI:SS')
简要说明,
YYYY 年
MM 月
- 【Scala六】分析Spark源代码总结的Scala语法四
bit1129
scala
1. apply语法
FileShuffleBlockManager中定义的类ShuffleFileGroup,定义:
private class ShuffleFileGroup(val shuffleId: Int, val fileId: Int, val files: Array[File]) {
...
def apply(bucketId
- Erlang中有意思的bug
bookjovi
erlang
代码中常有一些很搞笑的bug,如下面的一行代码被调用两次(Erlang beam)
commit f667e4a47b07b07ed035073b94d699ff5fe0ba9b
Author: Jovi Zhang <
[email protected]>
Date: Fri Dec 2 16:19:22 2011 +0100
erts:
- 移位打印10进制数转16进制-2008-08-18
ljy325
java基础
/**
* Description 移位打印10进制的16进制形式
* Creation Date 15-08-2008 9:00
* @author 卢俊宇
* @version 1.0
*
*/
public class PrintHex {
// 备选字符
static final char di
- 读《研磨设计模式》-代码笔记-组合模式
bylijinnan
java设计模式
声明: 本文只为方便我个人查阅和理解,详细的分析以及源代码请移步 原作者的博客http://chjavach.iteye.com/
import java.util.ArrayList;
import java.util.List;
abstract class Component {
public abstract void printStruct(Str
- 利用cmd命令将.class文件打包成jar
chenyu19891124
cmdjar
cmd命令打jar是如下实现:
在运行里输入cmd,利用cmd命令进入到本地的工作盘符。(如我的是D盘下的文件有此路径 D:\workspace\prpall\WEB-INF\classes)
现在是想把D:\workspace\prpall\WEB-INF\classes路径下所有的文件打包成prpall.jar。然后继续如下操作:
cd D: 回车
cd workspace/prpal
- [原创]JWFD v0.96 工作流系统二次开发包 for Eclipse 简要说明
comsci
eclipse设计模式算法工作swing
JWFD v0.96 工作流系统二次开发包 for Eclipse 简要说明
&nb
- SecureCRT右键粘贴的设置
daizj
secureCRT右键粘贴
一般都习惯鼠标右键自动粘贴的功能,对于SecureCRT6.7.5 ,这个功能也已经是默认配置了。
老版本的SecureCRT其实也有这个功能,只是不是默认设置,很多人不知道罢了。
菜单:
Options->Global Options ...->Terminal
右边有个Mouse的选项块。
Copy on Select
Paste on Right/Middle
- Linux 软链接和硬链接
dongwei_6688
linux
1.Linux链接概念Linux链接分两种,一种被称为硬链接(Hard Link),另一种被称为符号链接(Symbolic Link)。默认情况下,ln命令产生硬链接。
【硬连接】硬连接指通过索引节点来进行连接。在Linux的文件系统中,保存在磁盘分区中的文件不管是什么类型都给它分配一个编号,称为索引节点号(Inode Index)。在Linux中,多个文件名指向同一索引节点是存在的。一般这种连
- DIV底部自适应
dcj3sjt126com
JavaScript
<!DOCTYPE html PUBLIC "-//W3C//DTD XHTML 1.0 Transitional//EN" "http://www.w3.org/TR/xhtml1/DTD/xhtml1-transitional.dtd">
<html xmlns="http://www.w3.org/1999/xhtml&q
- Centos6.5使用yum安装mysql——快速上手必备
dcj3sjt126com
mysql
第1步、yum安装mysql
[root@stonex ~]# yum -y install mysql-server
安装结果:
Installed:
mysql-server.x86_64 0:5.1.73-3.el6_5 &nb
- 如何调试JDK源码
frank1234
jdk
相信各位小伙伴们跟我一样,想通过JDK源码来学习Java,比如collections包,java.util.concurrent包。
可惜的是sun提供的jdk并不能查看运行中的局部变量,需要重新编译一下rt.jar。
下面是编译jdk的具体步骤:
1.把C:\java\jdk1.6.0_26\sr
- Maximal Rectangle
hcx2013
max
Given a 2D binary matrix filled with 0's and 1's, find the largest rectangle containing all ones and return its area.
public class Solution {
public int maximalRectangle(char[][] matrix)
- Spring MVC测试框架详解——服务端测试
jinnianshilongnian
spring mvc test
随着RESTful Web Service的流行,测试对外的Service是否满足期望也变的必要的。从Spring 3.2开始Spring了Spring Web测试框架,如果版本低于3.2,请使用spring-test-mvc项目(合并到spring3.2中了)。
Spring MVC测试框架提供了对服务器端和客户端(基于RestTemplate的客户端)提供了支持。
&nbs
- Linux64位操作系统(CentOS6.6)上如何编译hadoop2.4.0
liyong0802
hadoop
一、准备编译软件
1.在官网下载jdk1.7、maven3.2.1、ant1.9.4,解压设置好环境变量就可以用。
环境变量设置如下:
(1)执行vim /etc/profile
(2)在文件尾部加入:
export JAVA_HOME=/home/spark/jdk1.7
export MAVEN_HOME=/ho
- StatusBar 字体白色
pangyulei
status
[[UIApplication sharedApplication] setStatusBarStyle:UIStatusBarStyleLightContent];
/*you'll also need to set UIViewControllerBasedStatusBarAppearance to NO in the plist file if you use this method
- 如何分析Java虚拟机死锁
sesame
javathreadoracle虚拟机jdbc
英文资料:
Thread Dump and Concurrency Locks
Thread dumps are very useful for diagnosing synchronization related problems such as deadlocks on object monitors. Ctrl-\ on Solaris/Linux or Ctrl-B
- 位运算简介及实用技巧(一):基础篇
tw_wangzhengquan
位运算
http://www.matrix67.com/blog/archives/263
去年年底写的关于位运算的日志是这个Blog里少数大受欢迎的文章之一,很多人都希望我能不断完善那篇文章。后来我看到了不少其它的资料,学习到了更多关于位运算的知识,有了重新整理位运算技巧的想法。从今天起我就开始写这一系列位运算讲解文章,与其说是原来那篇文章的follow-up,不如说是一个r
- jsearch的索引文件结构
yangshangchuan
搜索引擎jsearch全文检索信息检索word分词
jsearch是一个高性能的全文检索工具包,基于倒排索引,基于java8,类似于lucene,但更轻量级。
jsearch的索引文件结构定义如下:
1、一个词的索引由=分割的三部分组成: 第一部分是词 第二部分是这个词在多少

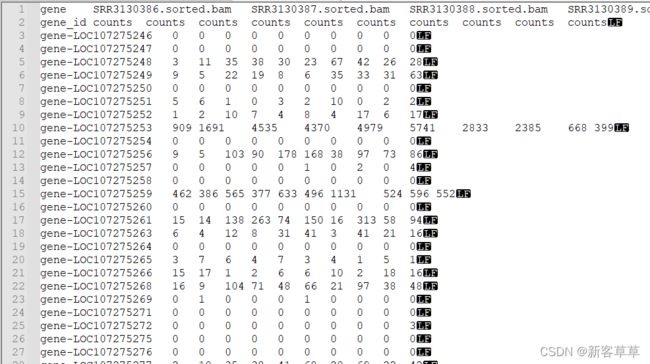 第一列表示基因的ID号,第一行表示样本的名称(一共有十个样本),矩阵中的数字表示该基因在该样本中的表达量
第一列表示基因的ID号,第一行表示样本的名称(一共有十个样本),矩阵中的数字表示该基因在该样本中的表达量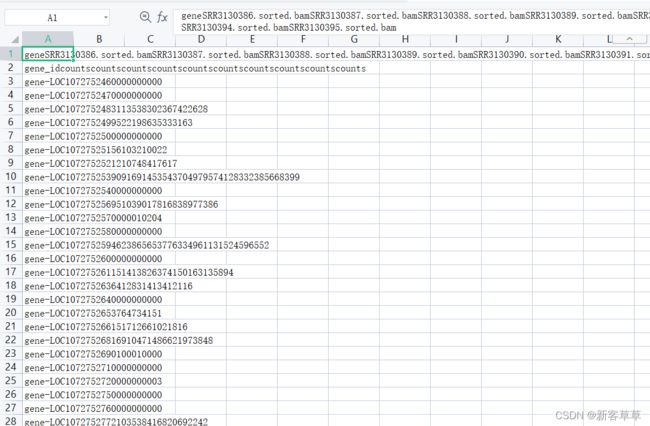 可以看出是把每行所有的数据放在一列了,按如下操作(选择所有行->数据->分列->完成)
可以看出是把每行所有的数据放在一列了,按如下操作(选择所有行->数据->分列->完成)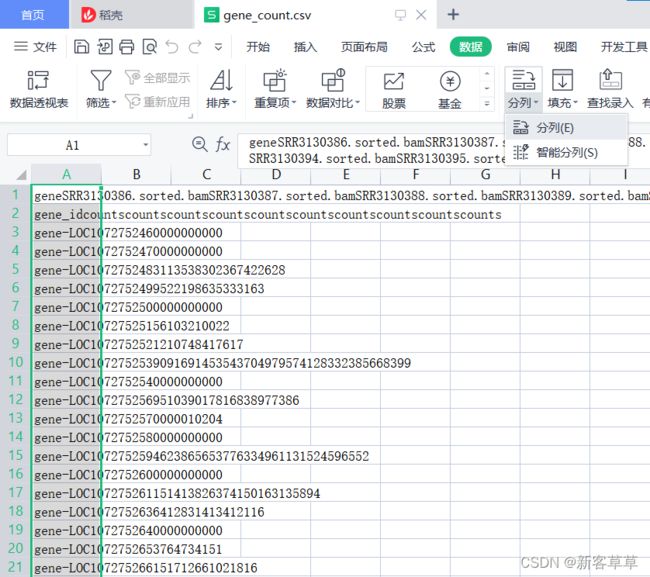
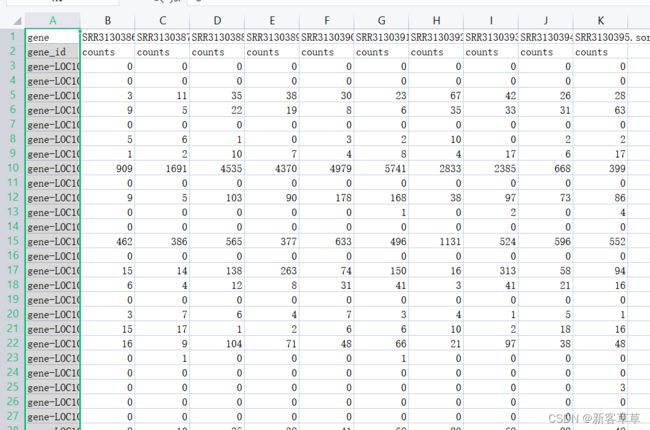 先把第二行删掉,因为有count字符会影响后续的操作
先把第二行删掉,因为有count字符会影响后续的操作 我决定用excel去做,在L2中输入下图所示公式(即计算中值绝对偏差)
我决定用excel去做,在L2中输入下图所示公式(即计算中值绝对偏差)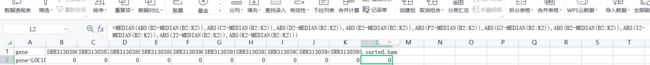 最后选中L列,按降序进行排序
最后选中L列,按降序进行排序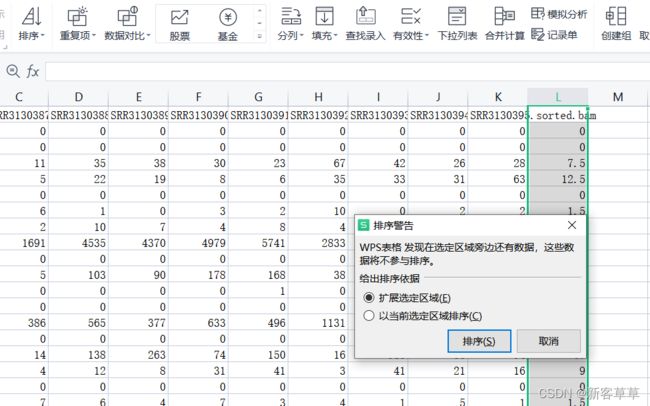 得到下图所示矩阵
得到下图所示矩阵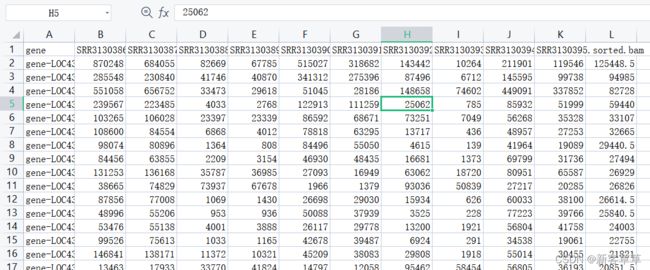 再复制其中前5000个数据,新建excel粘贴即可得到用于处理的表达矩阵,同时删除最后一列(因为最后一列存放的是中值绝对偏差),按下图操作,然后点击另存为,选择其他格式中的csv。千万不要直接改后缀,否则R语言读取时会出错的。
再复制其中前5000个数据,新建excel粘贴即可得到用于处理的表达矩阵,同时删除最后一列(因为最后一列存放的是中值绝对偏差),按下图操作,然后点击另存为,选择其他格式中的csv。千万不要直接改后缀,否则R语言读取时会出错的。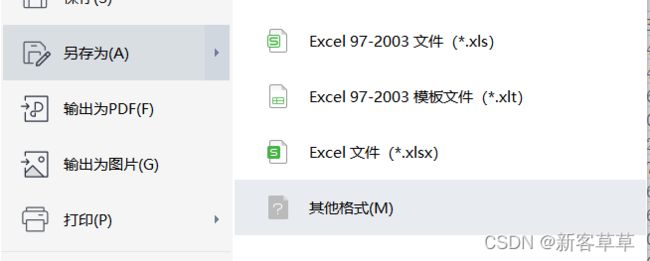

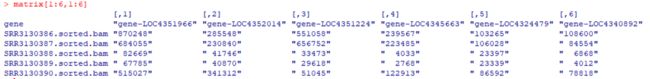 所以运行下面代码删除第一行
所以运行下面代码删除第一行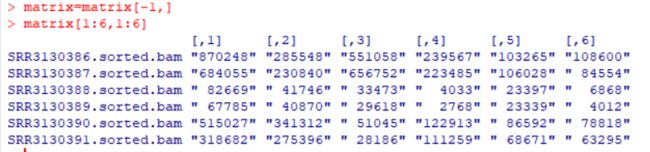
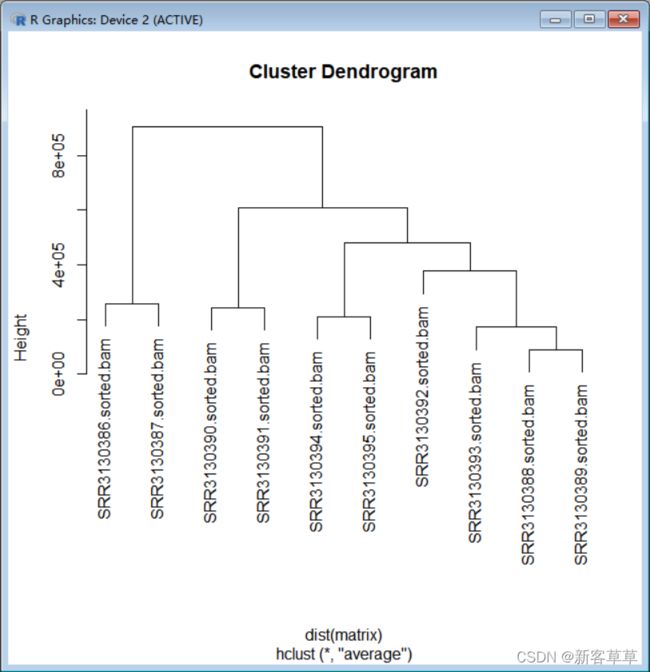 # 确定模型软阈值
# 确定模型软阈值