用R语言做WGCNA分析全步骤二(附代码解读)【转载】
代码逐句分析
- 一、文章来源
- 二、 关联模块与临床特征
-
- 1.量化module-trait(模块-特征)关系
- 2.网络可视化
一、文章来源
初学WGCNA,觉得博主的代码写的很不错,但其中很多代码在第一遍看的时候有很多地方不理解,后查阅了很多资料,终于看明白了,于是写了一篇笔记,记录自己的学习心得,有不准确的地方,还望各位大佬们不吝赐教~
原文链接:WGCNA简明指南
二、 关联模块与临床特征
1.量化module-trait(模块-特征)关系
在这个分析中,我们希望识别与临床特征显著相关的模块。由于我们已经有了每个模块的特征基因,我们简单地将特征基因与临床特征关联起来,并寻找最显著的关联。
# 导入之前的数据(也可以重新跑一遍上一期的内容),如果还没退出R,则不需要这一步
lnames =load(file="FemaleLiver-02-networkConstruction-auto.RData");
lnames
# 明确基因和样本数
nGenes =ncol(datExpr);
nSamples =nrow(datExpr);
# 用颜色标签重新计算MEs
# 按照模块计算每个module的MEs(也就是该模块的第一主成分)
# 按照下面的样式,得到的MEs0是将模块以颜色代表,各个样本中每个颜色的表达量,见下图
MEs0 = moduleEigengenes(datExpr, moduleColors)$eigengenes
# 对给定的(特征)向量重新排序,使相似的向量(通过相关性测量)彼此相邻。
MEs = orderMEs(MEs0) # 这时的MEs是将相似模块相邻后,即调整MEs0模块顺序后矩阵,这样在画模块性状关系图时,能够清晰捕捉特征
# 计算基因模块MEs 与 临床特征的相关性以及p值
# use 给出在缺少值时计算协方差的方法
moduleTraitCor =cor(MEs, datTraits, use ="p"); # 计算相关性系数
moduleTraitPvalue = corPvalueStudent(moduleTraitCor, nSamples) # 计算P值
MEs0矩阵的内容
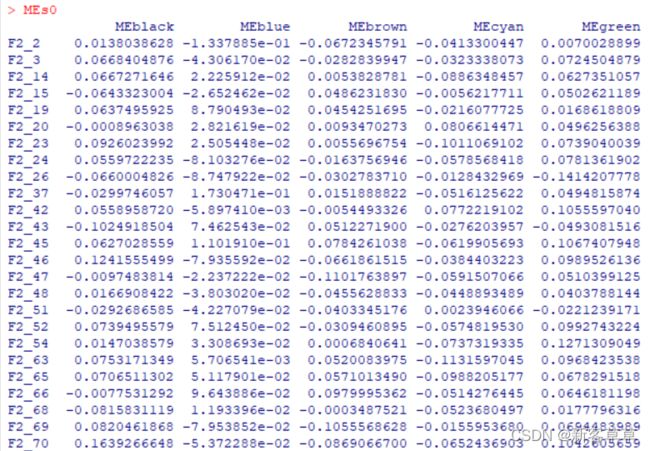
这时的MEs矩阵 ***这之后和原文提供的代码不一样,原文的代码有点问题。当然我们只要会用就行,将里面的参数进行修改即可。
***这之后和原文提供的代码不一样,原文的代码有点问题。当然我们只要会用就行,将里面的参数进行修改即可。
如果发现图片显示不全,修改代码中的par(mar=c(6,15,3,3))即可,这些分别代表下、左、上、右的页边距
可视化
# 如需保存
# pdf("Module-trait associations.pdf",width = 8, height=10)
par(mar =c(6, 15, 3, 3))
# 用热图展示相关性的结果
# 每个单元格标记相关系数和p值
textMatrix <- paste(
signif(moduleTraitCor, 2),
"\n(",
signif(moduleTraitPvalue, 1), ")",
sep = "")
dim(textMatrix) <- dim(moduleTraitCor)
labeledHeatmap(
Matrix = moduleTraitCor,
xLabels = names(datTraits),
yLabels = names(MEs),
ySymbols = names(MEs),
colorLabels = FALSE,
colors = greenWhiteRed(50),
textMatrix = textMatrix,
setStdMargins = FALSE,
cex.text = 0.5,
zlim = c(-1,1),
main = paste("Module-trait relationships"))
得到的结果如下图。每个单元格的颜色由对应的相关系数进行映射,数值从从-1到1,颜色由绿色过渡到白色,然后过渡到红色。
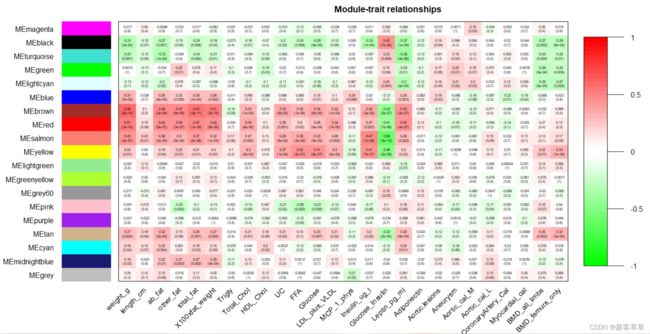 如果你是红绿色盲,那么可以不用绿色到红色,可以使用蓝白红的渐变。
如果你是红绿色盲,那么可以不用绿色到红色,可以使用蓝白红的渐变。
将上面的greenWhiteRed替换为blueWhiteRed,那么你将得到如下图
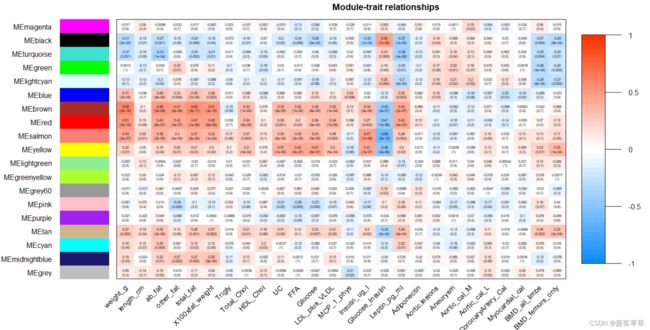 这个图我们怎么看(以蓝白红为例),矩阵上面的数据是相关系数,下面的即括号内数据是p值。通过上图可以看出每个基因模块与每个样本中的性状的关系(颜色越红即相关系数越高,说明关联性越强)。P值越小则置信度越高(置信度=1-P)
这个图我们怎么看(以蓝白红为例),矩阵上面的数据是相关系数,下面的即括号内数据是p值。通过上图可以看出每个基因模块与每个样本中的性状的关系(颜色越红即相关系数越高,说明关联性越强)。P值越小则置信度越高(置信度=1-P)
2.网络可视化
输入:getwd() 可得到我们在R语言中导出文件的存储位置
下面提到的矩阵等都是前面产生的,照本宣科即可
TOM = TOMsimilarityFromExpr ( datExpr, power = 6)
modules = c("brown","red")
probes = names (datExpr)
inModule = is.finite(match( moduleColors, modules));
modProbes = probes [ inModule];
modTOM = TOM[ inModule,inModule] ;
dimnames (modTOM) = list (modProbes,modProbes)
cyt = exportNetworkToCytoscape (
modTOM,
edgeFile = paste("CytoscapeInput-edges-",paste(modules,collapse="-"),
". txt", sep=""),
nodeFile = paste("CytoscapeInput-nodes-",paste(modules,collapse="-"),
". txt", sep=""),
weighted = TRUE,
threshold = 0.02,
nodeNames = modProbes ,
nodeAttr = moduleColors[inModule]
)
执行完后会得到两个文件,分别是CytoscapeInput-edges-brown-red.txt和
CytoscapeInput-nodes-brown-red.txt
将这个两个文件导入cytoscape后便可得到共表达网络图(这里由于结点太多,所以在这之前最好处理一下,否则图片非常不清晰)
参考方法:
①方法一
②方法二
③方法三