论文解读:《CoaDTI:基于多模态共同注意的药物-靶标相互作用注释框架》
、
论文解读《CoaDTI: multi-modal co-attention based framework for drug–target interaction annotation》
- 1.文章概述
- 2.知识点补充
-
- 2.1 GraphSAGE
- 2.2 视觉问答(Visual Question Answering)
- 2.3 r-radius 子图方法
- 3.背景
- 4.数据
-
- 4.1 人类数据集和秀丽隐杆线虫数据集
- 4.2 BindingDB 数据集
- 5.方法
-
- 5.1 问题表述
- 5.2 药物和靶标表示
-
- 5.2.1 用于药物表示的 SageGraph
- 5.2.2 用于蛋白质表示的Transformer
- 5.3 深度共同关注模块
- 5.4 分类器
- 6.结果
-
- 6.1 在人类数据集和秀丽隐杆线虫数据集上的表现
- 6.2 BindingDB 数据集上的性能
- 6.3 新药和蛋白质鉴定
- 6.4 稀缺数据的表现
- 6.5 注意力解读
- 7.关键点
文章地址:https://academic.oup.com/bib/advance-article-abstract/doi/10.1093/bib/bbac446/6770087?redirectedFrom=fulltext#supplementary-data
DOI:https://doi.org/10.1093/bib/bbac446
期刊:BRIEFINGS IN BIOINFORMATICS
2022年影响因子/JCR分区:13.994/Q2
出版日期:2022 10 月 23日
代码:https://github.com/Layne-Huang/CoaDTI
1.文章概述
- 动机:药物-靶标相互作用 (drug–target interactions,DTI) 的鉴定对于计算机药物发现起着至关重要的作用,其中药物是化学分子,目标是结合口袋(binding pockets)中的蛋白质残基。手动 DTI 注释方法虽然可靠,但测试每个药物-靶标对费力耗时。带标签的 DTI 数据的快速增长激发了人们对高通量 DTI 预测的兴趣。但这些方法高度依赖于人工表示的手动特征,从而导致错误。
- 结果:作者基于端到端的深度学习框架开发了一个名为 CoaDTI 的方法,用来提高药物靶标注释的效率和可解释性。 CoaDTI 结合了共同注意机制模拟来自药物模态和蛋白质模态的相互作用信息。特别的,CoaDTI 结合了 transformer 从原始氨基酸序列中学习蛋白质表示,并结合了 GraphSAGE 从 SMILES 中提取分子图特征。此外,作者建议采用迁移学习策略通过预训练的 transformer 对蛋白质特征进行编码,以解决标记数据稀缺的问题。实验结果表明,与最先进的模型相比,CoaDTI 在三个公共数据集上取得了有竞争力的性能。此外,迁移学习策略进一步提升了性能。扩展研究表明,CoaDTI 可以识别新的 DTI,例如候选药物与严重急性呼吸系统综合症冠状病毒 2 (severe acute respiratory syndrome coronavirus 2,SARS-CoV-2)相关蛋白之间的反应。共同注意分数的可视化可以说明模型对机械洞察力的可解释性。
2.知识点补充
2.1 GraphSAGE
GraphSAGE 是Graph SAmple and aggreGatE的缩写,其运行流程如上图所示,可以分为三个步骤:
- 对图中每个顶点邻居顶点进行采样;
- 根据聚合函数聚合邻居顶点蕴含的信息;
- 得到图中各顶点的向量表示供下游任务使用。
2.2 视觉问答(Visual Question Answering)
视觉问答任务是给定一张图片和一个与图片相关的问题,视觉问答模型来预测答案。视觉问答有很广泛的应用场景,例如早期教育、帮助盲人获取外部信息等。
2.3 r-radius 子图方法
r-radius 子图方法:Fast Neighborhood Subgraph Pairwise Distance Kernel
3.背景
药物-靶标相互作用 (DTI) 注释是药物开发的一项基本任务。揭示新的蛋白质-配体结合位点有助于确定潜在的副作用、毒性和药物重新定位。
最先进的 (state-of-the-art,SOTA) 方法,包括结构方法和机器学习方法,旨在自动准确地预测 DTI 的亲和力,为药物开发提供潜在的 DTI 候选者。结构方法利用考虑化学相互作用亲和力的分子对接来预测 DTI 。要计算相应的亲和力,需要高质量的三维 (3D) 信息。然而,蛋白质-配体对的共晶结构很难被研究人员确定,而高维结构矩阵总是稀疏的,导致大量繁琐和冗余的计算。
标记的 DTI 数据,尤其是药物-靶标结构数据的快速增长,引起了人们对采用机器学习方法进行 DTI 预测的兴趣。 DTI 预测任务被认为是一个二元分类任务,它集成了配体和目标特征以提取潜在信息,表明它们的相互作用。以前的研究试图利用包括化学和生物学描述符在内的高级特征来捕获交互表示。但这些方法严重依赖于手动特征,并且无法进行生物学解释。为了解决这个问题,作者直接关注低级信息,而放弃辅助功能。在这样的 DTI 任务中,只需要将一维蛋白质序列输入模型。隐藏在序列数据中的长短依赖关系可以通过递归神经网络 (RNN) 捕获,包括门递归单元 (GRU)和长短期记忆 (LSTM)。最近,transformer 已成为一种有前途且稳健的模型,通过利用自注意力来学习全局和局部序列依赖性来学习类序列数据的语义信息。此外,它还在图像等其他模态数据上取得了令人鼓舞的性能。图像数据需要展平以适应 transformer 的结构,而蛋白质原始数据是自然排序的。因此,很自然地使用 transformer 来捕获蛋白质序列的特征。
药物分子也可以表示为线性符号,通过使用短的 ASCII 字符串来注释其注释结构。具体来说,一个分子序列的长度总是小于100个重原子,这意味着它的结构空间相对较小。由于它可以表示为序列形式,因此 RNN 网络族适用于捕获底层特征。类似于使用 textCNN 提取单词序列的语义学习的 NLP 任务,卷积神经网络 (CNN) 结构已被用来挖掘分子序列的隐藏局部模式。此外,图神经网络 (GNN)已广泛应用于药物发现,包括 DTI 、分子特性估计和合成。由边和节点组成的图被输入到 GNN 模型中。在 DTI 预测问题中,化合物的原子和化学键被表示为节点和边,可以从药物分子序列中提取出来。与上述机器学习模型相比,GNN 在表示分子方面更加直观,因为它可以通过引入归纳偏差来整合结构信息,增强学习潜在空间功能相关性的能力。传统的 GNN 仅提取单个节点表示。相反,GraphSage 可以利用其适用于未见节点的邻域信息来生成节点嵌入。新型药物分子可能具有未包含在训练数据集中的不同结构。结合 GraphSage 可以使模型识别看不见的分子结构。
蛋白质和药物的表示可以看作是不同的数据模式。因此,为多模式任务设计的模型具有提升性能的潜力。以前的研究已经连接了蛋白质和药物分子的表示,并将连接的向量馈送到完全连接的层中。然而,单一的串联过程无法估计药物化合物的哪一部分对与靶蛋白的相互作用贡献最大,而忽略了交叉模态的互补性。自从首次针对视觉问答 (visual-question-answering,VQA) 任务提出多模态信息融合的注意机制以来,深度神经网络中的注意力机制在单模态任务和多模态任务中都取得了巨大成功。已经证明,共同注意可以学习图像和问题的细粒度表示。尽管研究人员采用单一注意力层来提取学习向量的相互作用,但很少有人尝试将深度同步学习和共同注意力应用于 DTI 问题。
为了解决这些问题,作者基于端到端的深度学习框架开发了一个名为 CoaDTI 的方法,用于直接基于低级多模态表示和深度共同注意机制来预测 DTI。输入的蛋白质序列被输入 transformer 以获得氨基酸水平的全局表示,而药物分子的表示是通过将 SMILES 序列输入 GraphSage 来学习的。受 VQA 任务中共同注意机制的启发,作者改进了自我注意机制来模拟模态内交互,并改进了共同注意来模拟模态间交互。迁移学习是从外部知识丰富数据特征的一个很有前途的方向。因此,将蛋白质模态中的 transformer 替换为预训练的 transformer 以规避标记数据的稀缺性,并为简洁起见将其注释为 CoaDTI-pro。作者进行了广泛的实验,以比较 CoaDTI 和 CoaDTI-pro 在具有真实数据设置的各种数据集上的性能。实验结果表明 CoaDTI 优于其他 SOTA 模型。特别是 CoaDTI-pro 将性能进一步提升到前所未有的水平。最后,蛋白质和药物交叉注意力得分的可视化使研究人员能够识别特定蛋白质-药物相互作用中的重要残基和原子,为下游制药任务提供有意义的线索。
4.数据
作者用不同的注意力模块对 CoaDTI 进行基准测试,用最合适的注意力模块(交叉点共同注意力)对 CoaDTI-pro 进行基准测试,并将性能与 SOTA 模型在人类数据集和线虫数据集上进行比较。此外,作者进一步将实验扩展到 BingdingDB 数据集上,以研究 CoaDTI 与真实数据设置的通用性和鲁棒性。
4.1 人类数据集和秀丽隐杆线虫数据集
人体数据集和秀丽隐杆线虫都包含通过系统筛选框架获得的化合物-蛋白质对的高度可信的阴性样本。假设与给定化合物的任何已知/预测目标不同的蛋白质不太可能被该化合物靶向,反之亦然,通过测量药物和蛋白质的距离得分来选择可靠的阴性样本通过蛋白质和药物的组合差异计算,而不是随机生成 DTI。为了公平比较,作者进行了平衡数据集,其中正样本和负样本的比例为 1:1。平衡的人类数据集包含与 1052 种独特化合物和 852 种独特蛋白质的 3369 种正相互作用。平衡的秀丽隐杆线虫数据集包含与 1434 种独特化合物和 2504 种独特蛋白质的4000 种正相互作用。80%的数据被划分为训练数据集,其余数据被认为是开发数据集(10%)和测试数据集(10%)。
4.2 BindingDB 数据集
BindingDB 数据集包含实验测量的结合亲和力。作者采用之前工作构建的数据集进行公平比较。该数据集包含 39 747 个正样本的相互作用和 31 218 个负样本的相互作用。 将数据集预先分为训练数据集(50155个相互作用)、验证数据集(5607个相互作用)和测试数据集(5508个相互作用)。

5.方法
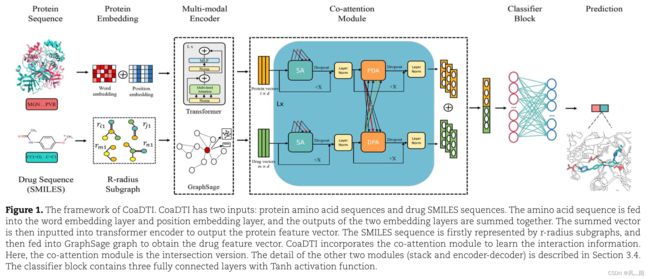
图 1 概述了所提出的基于多模态共同注意的框架 CoaDTI。给定药物 SMILES 序列的输入,CoaDTI 首先将线性序列转移到 R-radius 子图,这些子图是从顶点以 r 为半径导出邻近的顶点和边。子图数据被输入到 GraphSage 模型中,以利用邻域信息获得化合物的嵌入。考虑到残基的位置信息对于提取蛋白质特征至关重要,CoaDTI 采用两个嵌入层来获得具有不同频率的正弦和余弦函数的词嵌入和位置嵌入。 CoaDTI 结合了 transformer 的编码器来捕获包含隐藏全局信息的蛋白质表示。 vanilla transformer 进一步替换为预训练版本。在利用序列和图形表示后,CoaDTI 将信息从一种模态转换为另一种模态,并融合多模态信息以执行预测。为了将异构表示映射到公共空间,并估计药物化合物的哪一部分影响目标蛋白的贡献,CoaDTI 结合了深度共同注意机制来处理多模态特征。具体来说,CoaDTI利用self-attention分别对药物特征和蛋白质特征进行编码。然后这两个特征向量分别经过两个共同注意层,即DPA和PDA。 DPA 层查找药物对蛋白质的影响,而 PDA 层估计蛋白质对药物的注意力权重。作者还利用了不同的共同注意机制。串联全连接层用于将特征映射到交互概率。
5.1 问题表述
作者的模型用于预测药物与靶标的相互作用,这是一项二元分类任务。输入是药物-靶标对 (G, P),输出 y ∈ {0, 1},其中 y = 1 表示输入药物和输入蛋白质之间存在相互作用。
输入蛋白质表示为氨基酸序列 P = {a1, a2, . . . , an},其中 ai 是 23 种氨基酸的第 i 个索引。然后将蛋白质序列送入蛋白质嵌入层,将蛋白质序列映射到潜在特征空间。输入药物表示为包含结构信息的 SMILES 序列。通过找到化学键来提取原子的边缘,然后由边构造邻接矩阵。根据化学元素类型为原子分配索引。最后,得到化学结构图G = (v, ξ),其中v代表原子,ξ代表原子间的无向边,即化学键。
5.2 药物和靶标表示
5.2.1 用于药物表示的 SageGraph
药物分子表示为 SMILES(简化分子输入行输入系统)字符串。对于每个 SMILES 字符串,CoaDTI 从包含连接和结构信息的序列中提取原子和边缘信息。序列中的底层信息分布在非欧几里德空间中。归纳偏差使神经网络能够将结构赋予表征。为此,使用 GNN 通过信息传播提取高级节点表示是很直观的。直接将原子视为节点,将化学键视为边,是构建图形式数据的最明确方式。然而,分子图由几种类型的原子和化学键组成,导致图模型中的学习参数较少。因此,采用 r-radius 子图方法来表示复合图以解决该限制。


5.2.2 用于蛋白质表示的Transformer
考虑到蛋白质在 3D 空间中折叠反映了线性序列中的长距离依赖性,作者利用Transformer的编码器将蛋白质序列投影到密集的向量表示。
作为序列转导模型,Transformer 由两个嵌入层组成。第一个嵌入层是学习的嵌入层,它将输入标记转换为维度 dmodel = 512 的向量。第二个嵌入层是具有相同维度 dmodel 的位置嵌入层。然后采用不同频率的正弦和余弦函数来计算相对位置。然后将两个嵌入层的输出相加。对于每个输入的蛋白质序列,transformer 输出蛋白质表示 Xp ∈ Rl×d(l 是蛋白质序列的长度,d = 512)。
为了利用外部知识,作者用预训练的transformer 替换普通transformer ,并研究性能和鲁棒性的改进。预训练的 transformer 在 2.5 亿个蛋白质序列中的 860 亿个氨基酸上进行训练,跨越具有 4260 万个参数的进化多样性。
5.3 深度共同关注模块
作者采用注意力机制来整合来自蛋白质和药物形态的信息,以达到分类的目的。注意力机制的思想是利用有限的注意力资源,从海量数据中快速过滤掉高价值信息。图 2 展示了本研究中的三个注意力机制单元,自我注意力 (SA)、药物-蛋白质注意力 (DPA) 和蛋白质-药物注意力 (PDA)。类似于自然文本序列,蛋白质序列中的氨基酸和分子序列中的原子可能与其他氨基酸和原子有长或短的依赖关系。
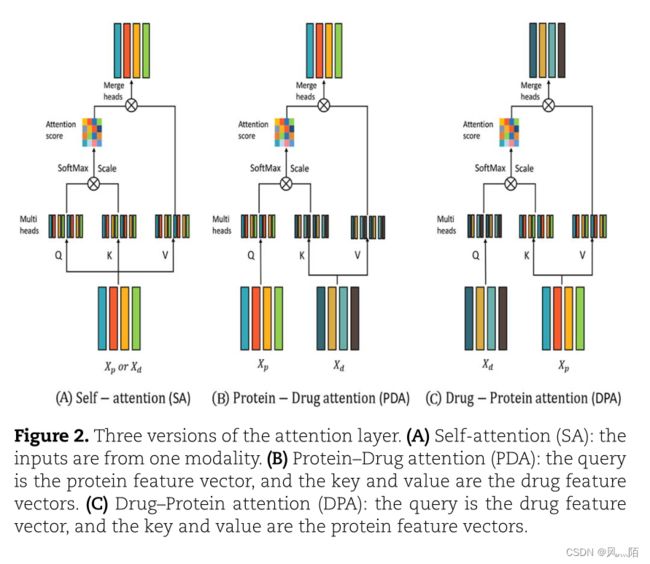
PDA 旨在研究药物分子的不同部分对靶蛋白的贡献。DPA 是测量蛋白质对药物的影响。
对于所有注意力单元,输出特征 Xupdate 被馈送到前馈层和丢弃层。结合了残差连接和层归一化,以进一步提高模块的鲁棒性。
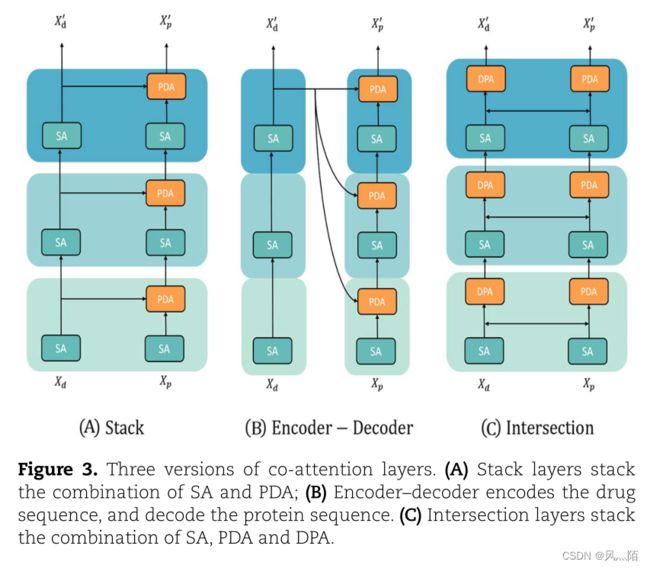
通过以不同的级联方式组装注意力单元来构建深度共同注意力模块(图 3)。堆栈模块(图 3A)堆栈 SA 和 PDA(SA,SA-PDA)的组合。 encoder-decoder模块(图3B)借鉴了sequence-sequence模型的思想,直观上适用于蛋白质和药物序列特征融合。编码器层通过堆叠 SA 层来学习药物序列特征,解码器层尝试通过堆叠 SAPDA 层将药物模态信息转换为蛋白质模态信息。交叉层(图 3C)旨在利用药物表示和蛋白质表示之间的相关性和相互作用。蛋白质和药物输入特征都首先由SA层输入。然后蛋白质特征由考虑药物效应的PDA层更新,而药物特征由考虑蛋白质效应的DPA层更新。输出分别为:X’p 和 X’d。
5.4 分类器
输出的X’p 和 X’d,在序列长度维度上进行平均以获得丰富的特征向量,然后再连接起来代表药物-靶点的特征向量。在分类块中,交互向量被馈送到具有激活函数 Tanh 的线性层。
6.结果
6.1 在人类数据集和秀丽隐杆线虫数据集上的表现
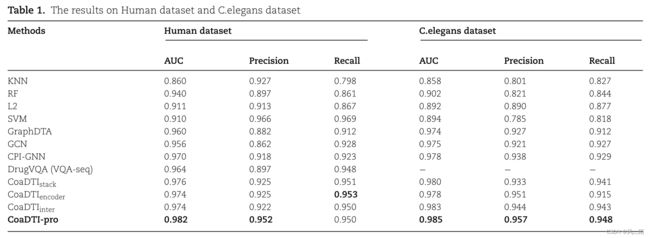
CoaDTIstack 是带有堆栈 co-attention 模块的模型。 CoaDTIencoder 是带有编码器-解码器co-attention 模块的模型。 CoaDTIinter 是带有交叉 co-attention 模块的模型。 CoaDTI-pro 利用预训练的 transformer 对蛋白质特征进行编码,并结合了带有交叉 co-attention 模块。
6.2 BindingDB 数据集上的性能
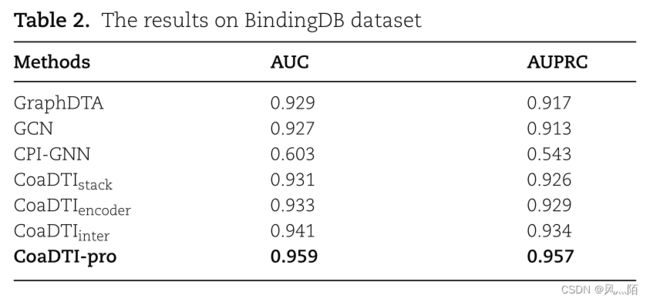
6.3 新药和蛋白质鉴定
选择 BindingDB 数据集作为广泛的数据集来评估 CoaDTI 的通用性和稳健性,因为它侧重于蛋白质和类药物小分子的相互作用。作者根据在训练数据集中是否观察到蛋白质或药物,将测试数据集进一步拆分为四个子集。最后,收集见过的药物和蛋白质子集,见过的蛋白质和看不见的药物子集,看不见的蛋白质和见过的药物子集和看不见的蛋白质和药物子集。将 CoaDTI-pro 与三个基线模型进行比较,包括基于相似性的方法 Tiresias、基于深度学习的方法 DBN,包括堆叠的受限玻尔兹曼机 (RBM) 和包含 LSTM 和 GCN 的端到端神经网络 E2E。

6.4 稀缺数据的表现
作者展示了 CoaDTI-pro 旨在通过利用预训练的 transformer 来规避正样本缺少的问题,该 transformer 已经学习了隐藏在大量未标记数据中的知识,如图 5 所示。

6.5 注意力解读
CoaDTI 中的共同注意模块可以使模型能够衡量药物对目标蛋白的影响以及蛋白质在相互作用中对药物的影响。作者选择了两个 SARS-CoV-2 3CL Mpro 相关相互作用:GC373(PDB ID:6WTK)和 ML188(PDB ID:7L0D)。注意力机制可以产生注意力权重来解释蛋白质特征向量和复合向量的焦点。
图 6 显示了 GC373 与 SARS-CoV-2 3CL Mpro 相互作用的化合物-蛋白质评分和蛋白质-化合物评分。特别是,化合物的原子会关注蛋白质序列(图 6a),而蛋白质载体则关注分子的特定原子(图 6b)。 SMILES序列顶部的原子所强调的区域比较平均,其他原子很可能会看位于蛋白质序列左侧的氨基酸。蛋白质序列集中在第9和第27位附近的原子上,呈现出整体关注的模式。

相互作用如图 7 所示,其中注意力得分高的氨基酸以红色突出显示,化合物的原子以绿色突出显示。蛋白质的注意力分数是通过在化合物维度上对蛋白质化合物注意力矩阵求平均来计算的,而化合物的注意力分数是通过在蛋白质维度上对化合物-蛋白质注意力矩阵进行平均来计算的。可以观察到,结合口袋内或周围的蛋白质残基和分子原子具有很高的注意力值。
7.关键点
- 提出了一个可解释的深度学习框架,以实现高通量药物-靶标结合注释,在 SOTA 模型中实现最佳性能,并为 SARS-COV-2 应用提供见解。
- CoaDTI 模型将药物和蛋白质视为两种不同的模式,可以从独立和共享的空间中学习信息。
- 该框架基于预训练的语言模型,用更少的训练数据将性能提升到前所未有的水平。
- CoaDTI 中的共同注意机制可以衡量药物和蛋白质的相互影响。研究人员可以通过使用 CoaDTI 阐明模型关注的地方来获得见解。