生信入门(一)——DESeq2差异基因分析
生信(一)——DESeq2差异基因分析
文章目录
- 生信(一)——DESeq2差异基因分析
-
-
- 一、差异基因分析原理
- 二、代码实现
-
-
- 1、前提:安装DESeq2包
- 2.代码实现
-
- 三、小结
-
记录学习过程,共勉。
一、差异基因分析原理
详见
二、代码实现
1、前提:安装DESeq2包

2.代码实现
setwd("D:\\RData");#设置编码位置
rt<-read.table("GSE149549_mRNA_Expression_Summary.txt",head=TRUE,row.names = 1)
head(rt)
#准备
rt<-as.matrix(rt) #将数据转换为矩阵格式
condition<-factor(c(rep("case",2),rep("control",3)),levels = c("case","control")) ## 设置分组信息,建立环境(5个样本,2组处理)
condition
coldata<-data.frame(row.names=colnames(rt),condition)
#剔除无意义数据(不存在的基因read)
#nrow(rt)
i=1
while(i<=nrow(rt)){
if(mean(rt[i,])<10)
rt<-rt[-i,]
i<-i+1;
};
#nrow(rt)
coldata #展示coldata内容
condition #展示环境
#构建dds矩阵
dds<-DESeqDataSetFromMatrix(rt,coldata,design=~condition)
#标准化,对原始数据进行normalize
dds<-DESeq(dds)
dds
resultsNames(dds)
#使用deseq2包中的result()函数提取deseq2差异分析结果
res=results(dds,contrast = c("condition","case","control"))
head(res)
summary(res)
write.csv(res,file = "results.csv")
#分析结果可视化与导出
table(res$padj<0.05)
plotMA(res,ylim=c(-2,2))
mcols(res,use.names = TRUE)
#提取差异基因
res <- res[order(res$padj),]
resdata <-merge(as.data.frame(res),as.data.frame(counts(dds,normalize=TRUE)),by="row.names",sort=FALSE)
deseq_res<-data.frame(resdata)
up_diff_result<-subset(deseq_res,padj < 0.05 & (log2FoldChange > 1)) #提取上调差异表达基因
down_diff_result<-subset(deseq_res,padj < 0.05 & (log2FoldChange < -1)) #提取下调差异表达基因
write.csv(up_diff_result,"up_case_VS_control_diff_results.csv") #输出上调基因
write.csv(down_diff_result,"down_case_VS_control_diff_results.csv") #输出下调基因
#plot(res$log2FoldChange,res$padj)#绘制火山图
注:DESeq2的表达数据必须是read counts,不能使用标准化后的FPKM 或TPM

由图可见,表达量(基因的样本平均count)数值越大,离散度越小,差异基因倍数越少
#提取两种算法标准化后的表达量(VST和rlog,大样本通常使用VST,推荐使用)
vsd<-vst(dds,blind = FALSE)
rld<-rlog(dds,blind = FALSE)
#对照使用 log 2(n+1)的标准化方法
ntd<-normTransform(dds)
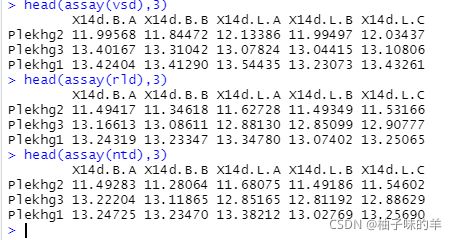
head(assay(vsd),3)
head(assay(rld),3)
head(assay(ntd),3)
#导出标准化后的数据(可用于GSEA等下游分析)
write.csv(as.data.frame(assay(vsd)),file="count_transformation_VST.csv")
write.csv(as.data.frame(assay(rld)),file="count_transformation_rlog.csv")
#绘制离散度散点图,离群值未做收缩
plotDispEsts(dds)

#对标准化后的数据进行可视化处理
#安装vsn包
#library(BiocManager)
#BiocManager::install("vsn")
#load package
#library("vsn")
#library(ggplot2)
#library(cowplot)
ntdl<-meanSdPlot(assay(ntd))
vsdl<-meanSdPlot(assay(vsd))
rldl<-meanSdPlot(assay(rld))
p1<-ntdl$gg+ggtitle("log2(n+1)")
p2<-vsdl$gg+ggtitle("VST")
p3<-rldl$gg+ggtitle("rlog")
plot_grid(p1,p2,p3,ncol = 3)
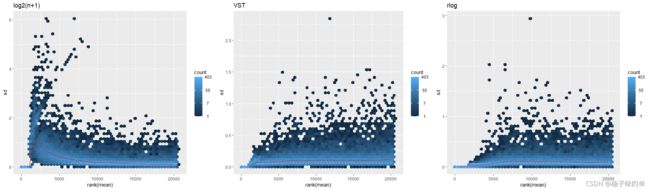
可见使用log2(n+1)标准化方法在低counts区域标准差较大,相反,rlog算法较大;而使用VST标准化后数据的标准差比较稳定;注意,虽然VST方法看起来效果最好,但图中的纵轴为方差的平方根(标准差),需要注意可能为由实验处理产生的真实方差。
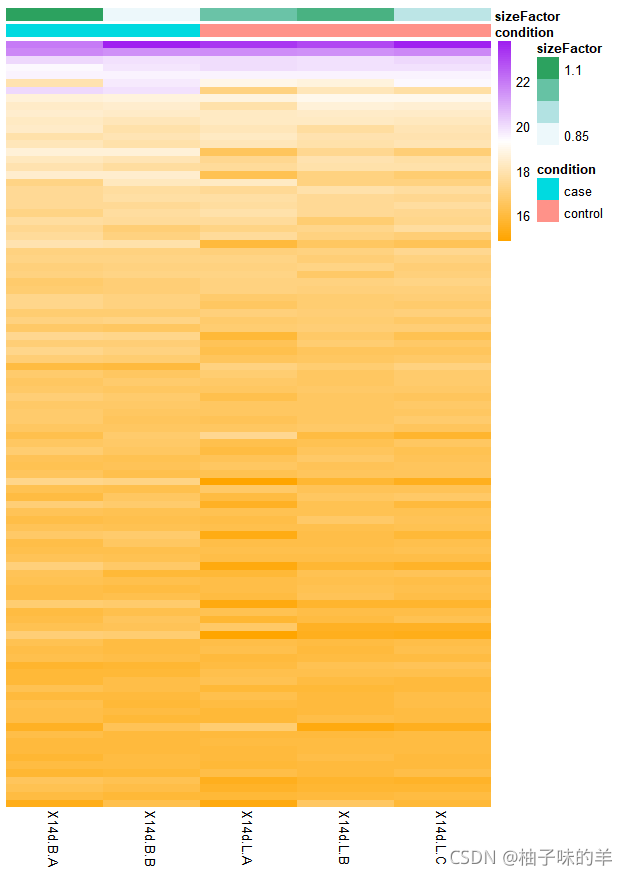
#使用标准化后平均表达量top100的基因绘制热图
library("pheatmap")
select<-order(rowMeans(counts(dds,normalized=TRUE)),decreasing = TRUE)[1:100]
colData(dds)
df<-as.data.frame(colData(dds)[,c("condition","sizeFactor")])
mycolors=colorRampPalette(c("orange","white","purple"))(100)
pheatmap(assay(vsd)[select,],cluster_rows=FALSE,show_rownames=FALSE,border_color=NA,color=mycolors,cluster_cols=FALSE,annotation_col=df)
#绘制样本距离聚类热图
#计算样本距离矩阵
sampleDists<-dist(t(assay(vsd)))
#由向量转成矩阵
sampleDistMatrix<-as.matrix(sampleDists)
rownames(sampleDistMatrix)<-paste(vsd$condition,vsd$sizeFactor,seq="-")
colnames(sampleDistMatrix)<-NULL
library("RColorBrewer")
colors<-colorRampPalette(rev(brewer.pal(8,"YlGnBu")))(255)
pheatmap(sampleDistMatrix,clustering_distance_rows = sampleDists,clustering_distance_cols = sampleDists,col=colors)

三、小结
刚入门,继续学习,加油!