数据挖掘—GEO,TCGA,Oncomine联合(三)GEO数据的下载和数据质量分析
系列文章目录
数据挖掘—GEO,TCGA,Oncomine联合数据(一)挖掘概述
数据挖掘—GEO,TCGA,Oncomine联合(二)GEO在线工具的应用
前言
要做分析那肯定要下载数据,这下载数据的过程大家肯定都会,但是下载完的数据真的能直接就使用吗?
使用工具: R
提示:以下是本篇文章正文内容,下面案例可供参考
一、数据质量分析有什么用吗?
GEO可以对芯片的数据进行管理,但是并不对数据的质量进行监控,所以这就需要我们对所下载的芯片的数据进行质量风险,规避之后做的分析有风险。
二、相对对数表达(RLE)与相对标准差(NUSE)
相对对数表达(RLE)一个探针组在对某个样品的表达值初一该探针组在所有样品中表达值得中位数取对数
相对标准差(NUSE):一个探针组在某个样品的PM值得标准差除以该探针组在各样品中的PM值标准差的中位数后取对数,反映了平行试验的一致性,比RLE更为敏感
三、步骤
数据下载大家肯定都会了吧,那我就直接说这个质量分析了,首先直接运行R语言就好,代码如下:
分析前准备
source("http://bioconductor.org/biocLite.R")
biocLite("affy")
biocLite("affyPLM")
biocLite("RColorBrewer")
biocLite("impute")
biocLite("limma")
biocLite("pheatmap")
install.packages("ggplot2")
setwd("") #自己设定
#调用R包
library(affyPLM)
#载入数据
Data<-ReadAffy()
#对数据集进行回归计算
Pset<-fitPLM (Data)
质量控制:查看灰度图
image(Data[,1])
#根据计算结果,画权重图
image(Pset,type="weights",which=1,main="Weights")
#根据计算结果,画残差图
image(Pset,type="resids",which=1,main="Residuals")
#根据计算结果,画残差符号图
image(Pset,type="sign.resids",which=1,main="Residuals.sign")
质量控制:相对对数表达(RLE)
一个探针组在某个样品的表达值除以该探针组在所有样品中表达之的中位数后取对数
反映平行实验的一致性
#调用R包
library(affyPLM)
library(RColorBrewer)
#对数据集进行回归计算
Pset<-fitPLM (Data)
#载入颜色
colors<-brewer.pal(12,"Set3")
#绘制RLE箱线图
Mbox(Pset,col=colors,main="RLE",las=3)
质量控制:相对标准差(NUSE)
一个探针组在某个样品的PM值的标准差除以该探针组在各样品中的PM值标准差的中位数后取对数。反映平行实验的一致性
比RLE更为敏感。
#调用R包
library(affyPLM)
library(RColorBrewer)
#对数据集进行回归计算
Pset<-fitPLM (Data)
#载入颜色
colors<-brewer.pal(12,"Set3")
#绘制NUSE箱线图
boxplot(Pset,col=colors,main="NUSE",las=3)
质量控制:RNA降解图
原理:RNA降解从5’端开始,因为芯片结果5’端荧光强度要远低于3’端
#调用R包
library(affy)
#获取降解数据
data.deg<-AffyRNAdeg(Data)
#绘制RNA降解图
plotAffyRNAdeg(data.deg,col=colors)
#在左上部位添加图注
legend("topleft",sampleNames(Data),col=colors,lwd=1,inset=0.05,cex=0.2)
RMA法预处理normal样本
setwd("")
library(affyPLM)
library(affy)
Data<-ReadAffy()
sampleNames(Data)
N=length(Data)
#用RMA预处理数据
eset.rma<-rma(Data)
#获取表达数据并输出到表格
normal_exprs<-exprs(eset.rma)
probeid<-rownames(normal_exprs)
normal_exprs<-cbind(probeid,normal_exprs)
write.table(normal_exprs,file="normal.expres.txt",sep='\t',quote=F,row.names=F)
RMA法预处理tumor样本
setwd("")
library(affyPLM)
library(affy)
Data<-ReadAffy()
sampleNames(Data)
N=length(Data)
#用RMA预处理数据
eset.rma<-rma(Data)
#获取表达数据并输出到表格
normal_exprs<-exprs(eset.rma)
probeid<-rownames(normal_exprs)
normal_exprs<-cbind(probeid,normal_exprs)
write.table(normal_exprs,file="tumor.expres.txt",sep='\t',quote=F,row.names=F)
合并N和T的数据
#setwd(" ")
normal_exprs<-read.table("normal.expres.txt",header=T,sep="\t")
tumor_exprs<-read.table("tumor.expres.txt",header=T,sep="\t")
#讲T和N合并
probe_exprs<-merge(normal_exprs,tumor_exprs,by="probeid")
write.table(probe_exprs,file="cancer.probeid.exprs.txt",sep='\t',quote=F,row.names=F)
Probe ID转换为Gene symbol(对平台文件进行整理)
setwd("")
#读取基因表达文件
probe_exp<-read.table("cancer.probeid.exprs.txt",header=T,sep="\t",row.names=1)
#读取探针文件
probeid_geneid<-read.table("GPL570-55999.txt",header=T,sep="\t")
probe_name<-rownames(probe_exp)
#probe进行匹配
loc<-match(probeid_geneid[,1],probe_name)
#确定能匹配上的probe表达值
probe_exp<-probe_exp[loc,]
#每个probeid应对的geneid
raw_geneid<-as.numeric(as.matrix(probeid_geneid[,3]))
#找出有geneid的probeid并建立索引
index<-which(!is.na(raw_geneid))
#提取有geneid的probe
geneid<-raw_geneid[index]
#找到每个geneid的表达值
exp_matrix<-probe_exp[index,]
geneidfactor<-factor(geneid)
#多个探针对应1个基因的情况,取平均值
gene_exp_matrix<-apply(exp_matrix,2,function(x) tapply(x,geneidfactor,mean))
#geneid作为行名
rownames(gene_exp_matrix)<-levels(geneidfactor)
geneid<-rownames(gene_exp_matrix)
gene_exp_matrix2<-cbind(geneid,gene_exp_matrix)
write.table(gene_exp_matrix2,file="Gastric.cancer.geneid.exprs.txt",sep='\t',quote=F,row.names=F)
#将gene id 转换为gene symbol
loc<-match(rownames(gene_exp_matrix),probeid_geneid[,3])
rownames(gene_exp_matrix)=probeid_geneid[loc,2]
genesymbol<-rownames(gene_exp_matrix)
gene_exp_matrix3<-cbind(genesymbol,gene_exp_matrix)
write.table(gene_exp_matrix3,file="Gastric.cancer.genesyb.exprs.txt",sep='\t',quote=F,row.names=F)
补充缺失值 (需要对genesyb这个文件需要处理)
最近邻居法(KNN,k-Nearest Neighbor)法:此方法是寻找和有缺失值的基因的表达谱相似的其他基因,通过这些基因的表达值(依照表达谱相似性加权)来填充缺失值
#调用函数impute
library(impute)
#读取表达值
gene_exp_matrix<-read.table("Gastric.cancer.genesyb.exprs.txt",header=T,sep="\t",row.names=1)
gene_exp_matrix<-as.matrix(gene_exp_matrix)
#KNN法计算缺失值
imputed_gene_exp<-impute.knn(gene_exp_matrix,k=10,rowmax=0.5,colmax=0.8,maxp=3000,rng.seed=362436069)
#读出经过缺失值处理的数据
GeneExp<-imputed_gene_exp$data
#写入表格
genesymbol<-rownames(GeneExp)
GeneExp<-cbind(genesymbol,GeneExp)
write.table(GeneExp,file="Gastric.cancer.gene.exprs.txt",sep='\t',quote=F,row.names=F)
library(limma)
rt<-read.table("Gastric.cancer.gene.exprs.txt",header=T,sep="\t",row.names="genesymbol")
#differential
class<-c(rep("normal",10),rep("tumor",10))
design<-model.matrix(~factor(class))
colnames(design)<-c("normal","tumor")
fit<-lmFit(rt,design)
fit2<-eBayes(fit)
allDiff=topTable(fit2,adjust='fdr',coef=2,number=200000)
write.table(allDiff,file="limmaTab.xls",sep="\t",quote=F)
#write table
diffLab<-allDiff[with(allDiff, ((logFC>1 |logFC<(-1)) & adj.P.Val<0.05)),]
write.table(diffLab,file="diffEXp.xls",sep="\t",quote=F)
共表达
diffExpLevel<-rt[rownames(diffLab),]
qvalue=allDiff[rownames(diffLab),]$adj.P.Val
diffExpQvalue=cbind(qvalue,diffExpLevel)
write.table(diffExpQvalue,file="diffExpLevel.xls",sep="\t",quote=F)
#heatmap
hmExp=log10(diffExpLevel+0.00001)
library('gplots')
hmMat=as.matrix(hmExp)
pdf(file="heatmap.pdf",height=120,width=90)
par(oma=c(3,3,3,5))
heatmap.2(hmMat,col='greenred',trace="none",cexCol=1)
dev.off()
#volcano
pdf(file="vol.pdf")
xMax=max(-log10(allDiff$adj.P.Val))
yMax=max(abs(allDiff$logFC))
plot(-log10(allDiff$adj.P.Val),allDiff$logFC,xlab="adj.P.Val",ylab="logFC",main="Volcano",xlim=c(0,xMax),ylim=c(-yMax,yMax),pch=20,cex=0.4)
diffSub=subset(allDiff,allDiff$adj.P.Val<0.05 & abs(allDiff$logFC)>1)
points(-log10(diffSub$adj.P.Val),diffSub$logFC,pch=20,col="red",cex=0.4)
abline(h=0,lty=2,lwd=3)
dev.off()
source("http://bioconductor.org/biocLite.R")
biocLite("clusterProfiler")
biocLite("pathview")
setwd("")
library("clusterProfiler")
rt=read.table("X.txt",sep="\t",head=T,check.names=F)
geneFC=rt$logFC
gene<-rt$ENTREZ_GENE_ID
names(geneFC)=gene
#kegg
kk<-enrichKEGG(gene=gene,organism="human",qvalueCutoff=0.05,readable=TRUE)
write.table(summary(kk),file="KEGG.xls",sep="\t",quote=F,row.names=F)
pdf(file="KEGG.barplot.pdf")
barplot(kk,drop=TRUE,showCategory=12)
pdf(file="KEGG.cnetplot.pdf")
cnetplot(kk,categorySize="geneNum",foldChange=geneFC)
library("pathview")
keggxls=read.table("KEGG.xls",sep="\t",header=T)
for(i in keggxls$ID){
pv.out<-pathview(gene.data=geneFC,pathway.id=i,species="hsa",out.suffix="pathview")}
大家一定要在setwd那行输入自己的工作目录啊!
四、结果

这四个图为一组图,第一个图为灰度图,一个合格的灰度图应该是大面积呈现灰色的图,如果在图中出现了一个白块那么就代表这个芯片数据存在风险,第二章为权重图,我这个比较模糊,但是正常的情况下,绿色的点代表权重较低,白色或者灰色代表权重较高,如果权重随机分布,那么绿色的点分布会比较均匀,第三个图为残差图,红色代表正的高残差白色代表正的低残差,蓝色代表负残差,点都是均匀分布在图中,如果第二张图和第三张图中的红色,绿色,蓝色点都是随机分布那么代表这个新品数据是合格的,第四张图红色的代表正残差,蓝色代表的是负反差,如果随机分布没有明显的同一颜色的点聚集在一起那么就代表数据是合格的。
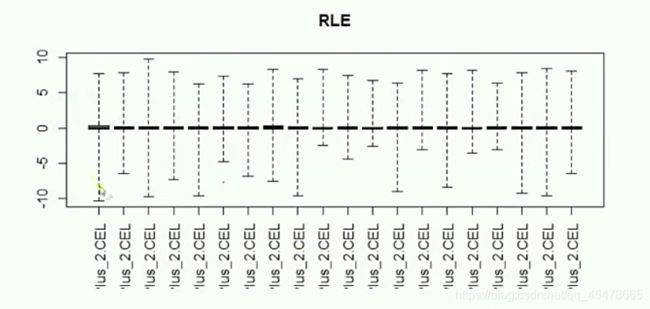
这个图可以看出这个芯片的数据质量,每个样本的中心线都很接近都很接近0,这个就代表这个芯片的数据是合格的,如果所有的数据也很接近,但是接近于5或者接近于10代表这个芯片的数据有问题,不利于做实验
这张图是相对标准差图,基本上所有

的样品大约都在一个水平线上,而且都接近于1这代表这个数据是可靠的,如果不处于一个水平线上而且还趋向于大于1的数那么很有可能是不可靠的。

这张图是RNA 降解图,RNA降解是影响芯片数据很关键的一个因素,RNA是从5‘端开始降解,所有5‘端比3’端低,这是正常的芯片数据评价,如果斜率处于低斜率或者是水平的,可能样品RNA已经全部被降解,如果斜率特别大,这就代表5‘端被降解的RNA过多这就代表数据有风险。
总结
总结
本章主要是讲解了GEO的数据的的质量分析,在用芯片数据做分析的时候,需要保证自己所下载的数据可以使用没有风险,才可以保证实验所做的结果有定的可信性。
下一章我会讲解(四)对于下载的原始数据进行处理
最后我所做的所有分析与教程的代码都会在我的个人公众号中,请打开微信搜索“生信学徒”进行关注,欢迎生信的研究人员和同学前来讨论分析。
ps:公众号刚刚建立比较简陋,但是该有的内容都不会少。图片有的很模糊,主要是因为我用的qq截图然后想直接放到文章里来,结果他不让,我只能放到桌面,在从桌面拖进来,像素就越来越低,生气!