limma 包的normalizeBetweenArrays和其他数据矫正方法removeBatchEffect
limma 包的normalizeBetweenArrays和其他数据矫正方法
2.normalizeBetweenArrays只能是在同一个数据集里面用来去除样本的差异,不同数据集需要用limma 的 removeBatchEffect函数 去除批次效应
原文链接:https://my.oschina.net/u/4503882/blog/4511789
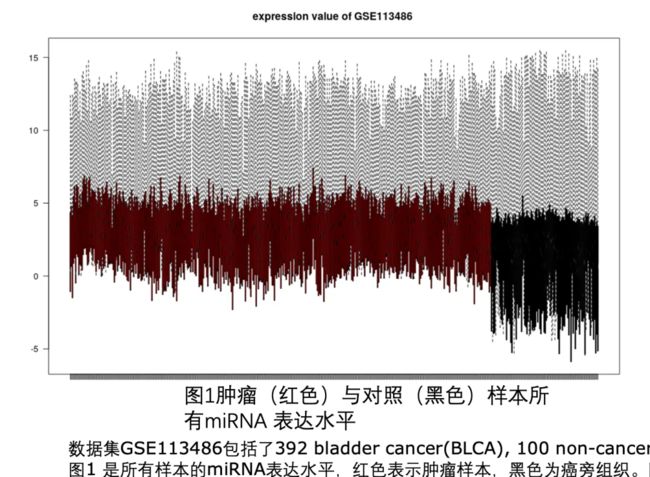
数据矫正前.png
可以看到,肿瘤样品的表达量整体就比正常对照样品的表达量高出一大截,这样的数据进行后续分析,就会出现大量的上调基因。
因为我一直强调,做表达矩阵分析一定要有三张图,见:你确定你的差异基因找对了吗? ,所以就让粉丝继续摸索,其中PCA如下:
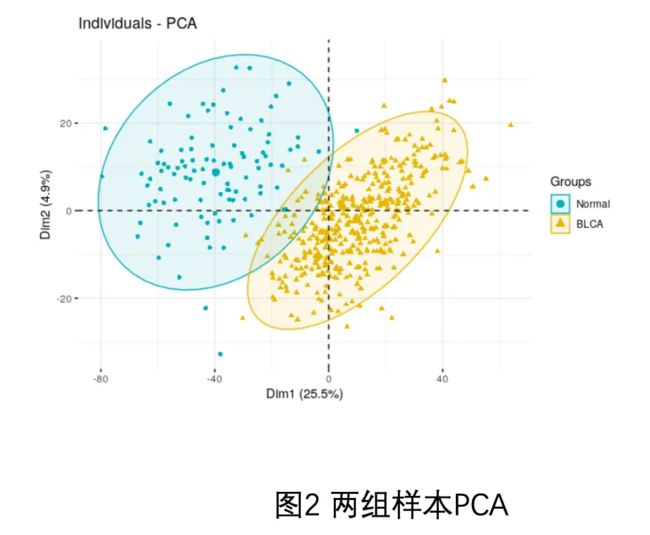
做表达矩阵分析一定要有三张图.png
可以看到,两个分组是泾渭分明的,这可能是生物学差异,因为肿瘤样品就肯定跟正常组织不一样的啊,也有可能是批次效应。所以我给粉丝的建议是两个策略
第一个策略是直接normalizeBetweenArrays处理,然后走差异分析。
第二是先去除批次效应,然后走差异分析。
建议你比较一下,这两个差异分析的区别。
然后粉丝的行动也很迅速,两三天就回复了邮件,给出了两个摸索结果:
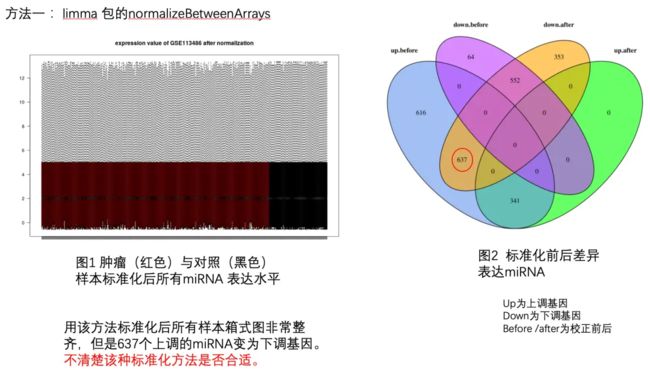
方法一:limma 包的normalizeBetweenArrays.png
可以看到,直接normalizeBetweenArrays处理其实就是一个quantile的normalization而已,大家可以去看quantile normalization到底对数据做了什么 - 简书,了解一下。
从韦恩图可以看到,没有进行normalizeBetweenArrays处理之前呢,上调基因真的是超级多啊!经过了normalizeBetweenArrays处理之后呢,其中616个上调基因变成了没有显著性改变的基因,然后637个居然由上调基因变成了下调基因,当然了,也有341个基因维持原来的上调属性。
是不是很可怕!!!
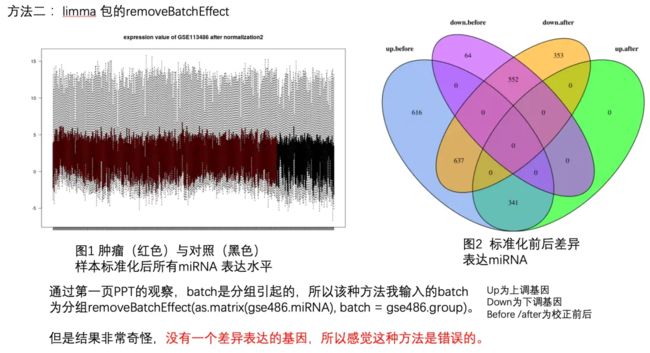
方法二:limma 包的removeBatchEffect.png
粉丝下的这个结论很正确,这个时候使用 limma 的 removeBatchEffect 函数来矫正批次效应,肯定是错的,因为完全没有搞清楚矫正批次效应的统计学原理。
其实几年前我在《单细胞天地》公众号发起过一个谈论,见:到底是批次效应还是真实生物学差异,如果你仅仅是做了两个单细胞转录组样品,想合并这两个数据再后续分析,就面临着两个样品(处理前后的生物学差异)本身的批次效应(不同时间点取样,不同10x上机时间等等)。因为是单细胞,一个样品里面本身就有这成千上万个细胞,可以针对两个样品内部的某些具有不变属性的单细胞来作为锚定,从而比较好的合并两个样品的单细胞转录组数据。
但是,如果是bulk转录组测序,或者表达量芯片,就基本上不可能做到区分具有生物学差异的两个样品的批次效应了。虽然说我在《生信技能树》写过不少相关教程,比如:多种批次效应去除的方法比较,但那样的去除是针对生物学差异与批次效应交叉的情况来去除。比如:
- 第一个批次:2个处理,2个对照样品
- 第二个批次:3个处理,3个对照样品
这个时候,就可以使用 limma 的 removeBatchEffect 函数或者 sva 的 ComBat 函数,把批次效应去除掉,然后保留生物学差异供后续的差异分析。
但是如果你的实验设计是:
- 第一个批次:3个处理样品
- 第二个批次:3个对照样品
那我就只能奉劝你,对这个数据集说拜拜了!
normalizeBetweenArrays
rt=read.table("lncRNA.txt",sep="\t",header=T,check.names=F)
#重复基因取均值,去重复
rt=as.matrix(rt)
rownames(rt)=rt[,1]
exp=rt[,2:ncol(rt)]
dimnames=list(rownames(exp),colnames(exp))
rt=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)
rt=avereps(rt)
rt=rt[rowMeans(rt)>0,]
#1个数据集批内矫正
rt=normalizeBetweenArrays(as.matrix(rt))
boxplot(rt)
removeBatchEffect
rt1=read.table("lncRNA1.txt",sep="\t",header=T,check.names=F)
rt2=read.table("lncRNA2.txt",sep="\t",header=T,check.names=F)
rt1=as.matrix(rt1)
rt2=as.matrix(rt2)
rt3<-merge(rt1,rt2)
rownames(rt3)=rt3[,1]
exp=rt3[,2:ncol(rt3)]
dimnames=list(rownames(exp),colnames(exp))
rt3=matrix(as.numeric(as.matrix(exp)),nrow=nrow(exp),dimnames=dimnames)
#查看未作批次矫正前的表达图
boxplot(rt3)
#多个数据集批次矫正
batch <- c(rep('con',182),rep('treat',32))
group_list <- c(rep('tumor',6),rep('normal',6),rep(c('tumor', 'normal'),each=3))
design=model.matrix(~group_list)
rt=removeBatchEffect(rt3,batch = batch,design = design)
boxplot(rt)
write.table(rt,file="rt.xls",sep="\t",quote=F)