【生信MOOC】生物序列比对工具——多序列比对
【生信MOOC】生物序列比对工具2——多序列比对
文章的文字/图片/代码部分/全部来源网络或学术论文,文章会持续修缮更新,仅供大家学习使用。
目录
【生信MOOC】生物序列比对工具2——多序列比对
1、多序列比对的定义和用途
2、多序列比对的要求
3、多序列比对工具——EMBL - Clustal Omega
4、多序列比对工具——EMBL - TCOFFEE - Expresso
5、多序列比对的保存格式
6、多序列比对结果编辑——jalview
7、寻找保守区域:序列标识图 WebLogo
8、寻找保守区域:序列基序 MEME
9、寻找保守区域:PRINTS 指纹图谱数据库
1、多序列比对的定义和用途
定义:两条以上的 生物序列进行的全局比对就是多序列比对。
用途:
1)可以通过多序列比对确定某一个未知序列是否属于某一个家族。
2)可以用多序列比对构建系统发生树,查看物种间或者序列间的进化关系。 做多序列比对是构建系统发生树的必要步骤之一。
3)模式识别。一些特别保守的序列片段 往往对应着重要的功能区。通过多序列比对,可以找到这些保守片段,并由此推测出潜在功能区。
4)可以把已知的有特殊功能的序列片段通过多序列比对做出匹配模型。然后根据这个模型推测未知的序列片段是否也具有这个功能。
5)多序列比对在生物信息学分析的很多方面都有应用,比如用来预测蛋白质的二级结构和三级结构,预测RNA 的二级结构等等。
两条序列的比对:需要构建一个二维表格,然后从 右下角到左上角找出一条最优路线。
如果是3 条序列的比对:应该做一个三维立方体,从 (0,0,0)这个位置到(n,n,n)这个位置找到最优的贯穿路径。
以此类推,做 n 条序列的比对:就要创建一个 n 维空间。这个 n 维空间实在是难以想象,但是有一点是明确的, 就是到了 n 维我们已经没有办法再像二维那样精确的计算出比对结果了。
由于计算量过于巨 大,所以目前所有的多序列比对工具都是不完美的。它们都使用一种近似的算法。目的就是 为了缩短计算时间,但也因此牺牲了一定的准确度。好在多序列比对并不像双序列比对对准 确度要求极高。通常,我们是要从多序列比对中看到一个趋势,一个大体的位置,所以牺牲掉的这点儿准确度影响不大。
2、多序列比对的要求
多序列比对的序列有一定要求:
1)做多序列比对的序列个数不能太多,一般 10 到 15 条序列刚好,最好不要超过 50 条。 序列太多,任何软件都受不了。
2)关系太远的序列不适合做多序列比对。两两之间序列相似度低于 30%的一组序列,做多序列比对要么做不出来,要么即使勉强做出来了,做得也是零七八碎,没有任何意义。
3)关系太近的序列不适合做多序列比对。两两之间序列相似度大于 90%的序列,有再多条都只等于一条。做出来的多序列比对无非就是把各条序列抄写了一遍,没有任何意义。
4)短序列受不了。多序列比对支持一组差不多长的序列,个别很短的序列纯属捣乱分子。
5)有重复域的序列受不了。如果序列里包含重复片段,大多数多序列比对的程序都会出错,甚至崩溃。
给序列起名字的要求:
1)序列名字里不要有“空格”,用下划线代替“空格”是个好习惯。
2)不要使用特殊字符,比如中文,@,#之类的。
3)序列名字不要太长,最好在 15 个字符以内。
4)一组序列里,不要有重名的序列。
3、多序列比对工具——EMBL - Clustal Omega
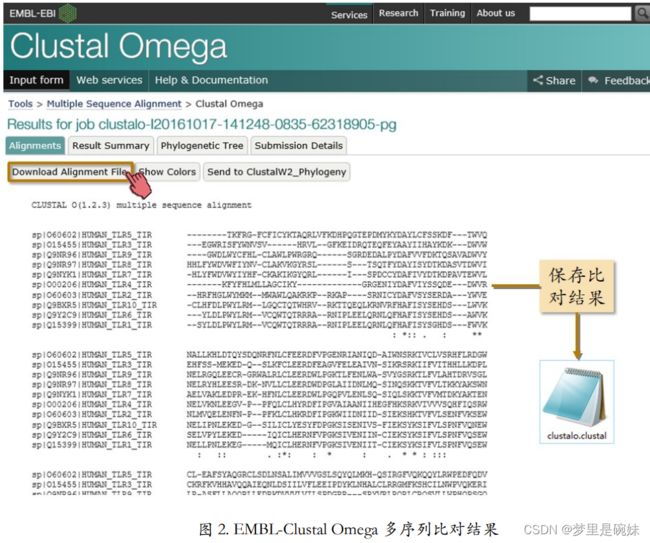
参数里有输出格式(OUTPUT FORMAT)和输出顺序(ORDER)这两个参数。输出格式里可以选择常用的多序列比对格式。我们选标准的 Clustal 格式。这是最常见的多序列比对格式。
输出顺序参数可以设定多序列比对中各个序列的排列顺序。“aligned” 是按照比对过程中自动创建的计算顺序排列;“input”是按照输入序列的原始顺序排列。输入序列是按照 TLR10、9、8、7…这样的顺序排列的。
如果某一列是完全保守的一列, 也就是说这一列里的字母完全相同,那么这一列下面就打一个“*”。
如果这一列的残基有大致相似的分子大小及相同的亲疏水性,也就是这一列的字母要么相同要么相似,没有不 相似的,那么就打一个“:”。
如果这一列残基的分子大小及亲疏水性被一定程度上保留了, 但是有替换发生在不相似的残基间,也就是这一列的字母有相似的也有不相似的,那么就打 一个“.”。
什么都不标记代表这一列是完全不保守的,也就是这一列的字母全部都不相似。
这些星星点点的标记可以为我们指出保守区域所在的位置,即星星点点特别密集的区域。
Result Summary 标签里,给出了全部结果信息的下载列表和一个 Jalview 的按钮(图 4)。 Jalview 是多序列比对编辑软件,在下载列表里,如果打开 “Percent Identity Matrix”链接,可以得到所有序列两两之间的一致度矩阵。一致度矩阵的第 一行省略掉了。它和第一列完全相同,都是序列的名字并且按照相同的顺序排列。所以这个 矩阵是以对角线为轴对称的,并且对角线上是某条序列自己和自己的一致度,都是100%。 这个矩阵可以帮助我们更好的了解这些序列之间的关系。
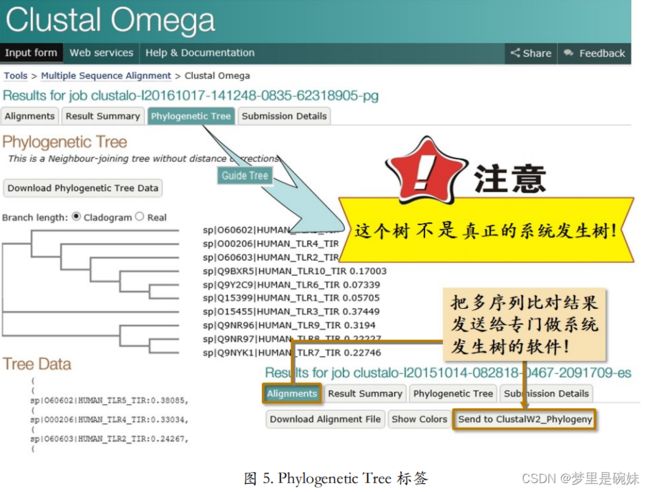
除了通过一致度矩阵了解序列间的关系,还可以通过 Phylogenetic Tree 标签下的 Guide Tree 清楚的看出哪条序列和哪条序列更相似。Phylogenetic Tree 翻译成中文是系统发生树。但是这里要特别注意,这不是真正意义上的系统发生树!它只是在创建多序列比对的 过程中用到的树(Guide Tree),没有经过距离校正,所以不能当作系统发生树来使用。如果 想要根据多序列比对结果构建系统发生树,可以在 Alignments 标签下,点击“Send to ClustalW2_Phylogeny”链接,把做好的多序列比对发送给专门做系统发生树的工具。
4、多序列比对工具——EMBL - TCOFFEE - Expresso
TCOFFEE 是一个非常流行的多序列比对工具。
TCOFFEE 与 CLUSTAL 系列在所使用 的算法上类似,准确度上比 CLUSTAL 系列略高,但计算耗时也比 CLUSTAL 系列略高。
最关键的是 TCOFFEE 有很多种变形,也就是说它有更多的功能。
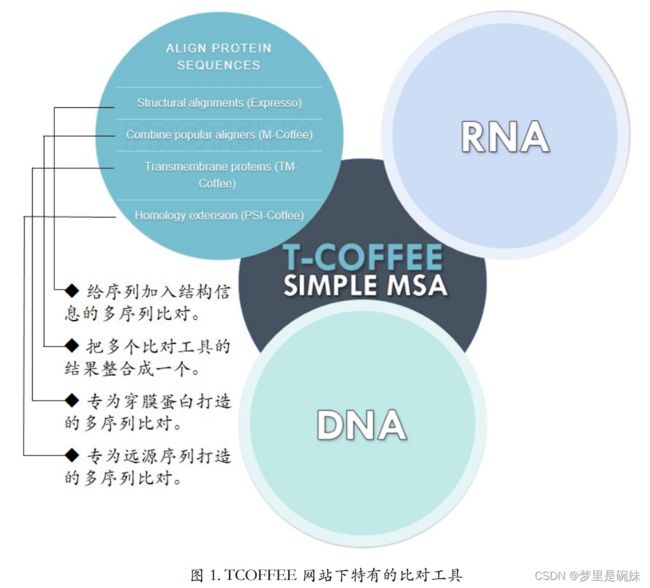
Expresso 是为序列加入结构信息后再做多序列比对的工具。因为有结构信息的辅助,它可以大大提高比对的准确度。
M-Coffee 可以把多个比对的结果整合成一个。TM-Coffee 专为穿膜蛋白打造。
PSI-Coffee 专为远源序列打造。同样的还有针对 RNA 和 DNA 序列的 Coffee。
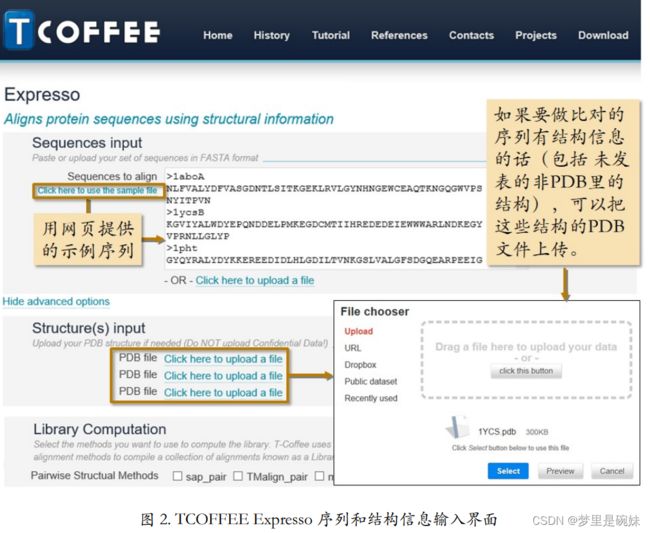
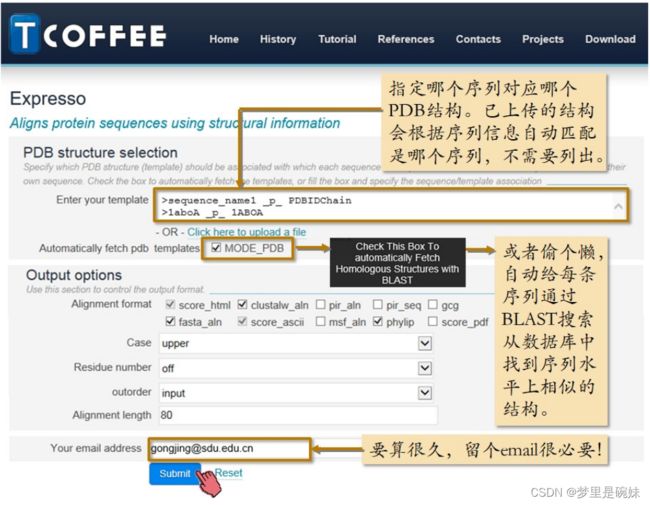
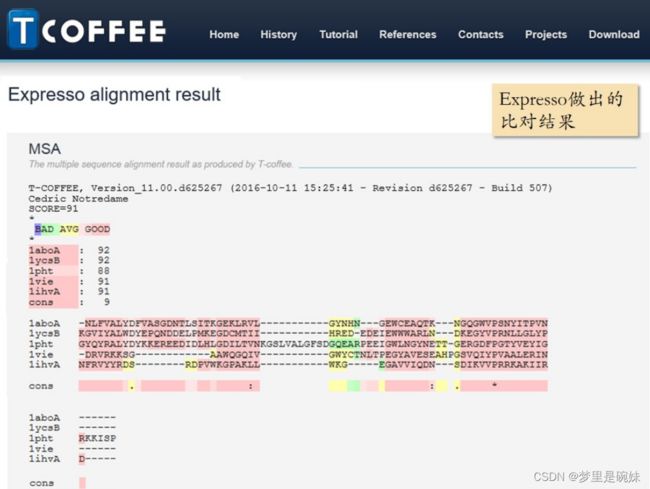
同样的序列做普通的 TCOFFEE,质量远不如 Expresso。可以看到二级结构全部 错位。所以,如果你有序列的结构信息的话,用 Expresso 相比用普通的比对工具会大大提高比对质量。
5、多序列比对的保存格式
在选择保存格式之前,需要问自己几个问题:
1)你选的这个格式大多数软件都支持吗?
2)你的同事能用吗?
3)你需要的信息这个格式都提供了吗?
4)这个格式适合进一步加工吗?
也可以通过第三方软件进行格式转换。比 如 fmtseq 工具(http://www.bioinformatics.org/JaMBW/1/2),它可以实现 20 多种格式间的转换。
6、多序列比对结果编辑——jalview
快速版的jalview,在 EMBL Clustal Omega 比对结果的 Result Summary 标签下 有 Jalview 按钮。这个按钮可以快速启动 Jalview,但这里启动的在线版本功能不完整。
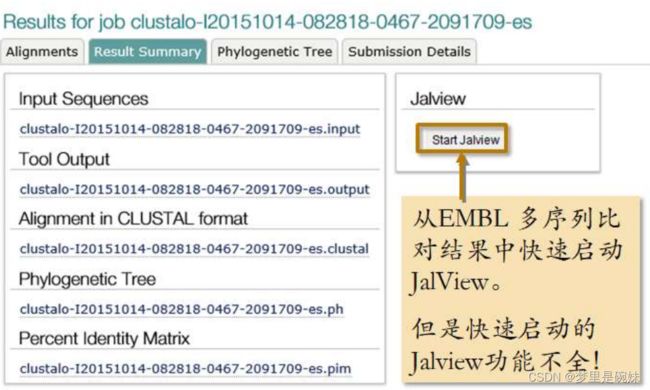
完全版的 jalview 可以从 Jalview 官网(http://www.jalview.org)在线启动,或者下载安装 到本地。
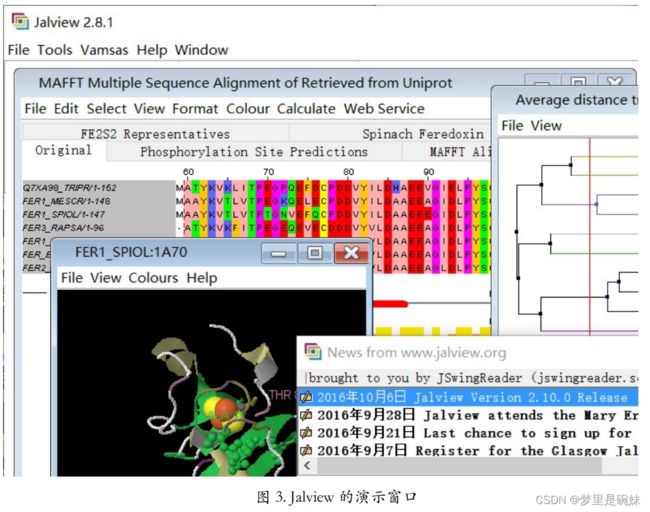
在打开的多序列比对窗口的下方有三行柱状图。它们体现了比对中每个位置的 保守度高低(Conservation)、比对质量高低(Score)、以及共有序列(Consensus)。从保守度 行,可以很清楚的找到保守区大致的位置。
共有序列指的是某一列出现频率最高的那个字母, 比如第 58 列中 W 出现的频率最高,是 100%。如果某一列拥有的最高出现频率的字母是两 个或两个以上的话,会以“+”显示。把鼠标放在“+”上就可以看到是哪些字母出现的频 率一样高。共有序列可以一定程度上体现出某个保守区域所具有的序列特征。以后如果看到 和这段序列长相极其相似的序列,它很可能能跟这个保守区的功能相似。
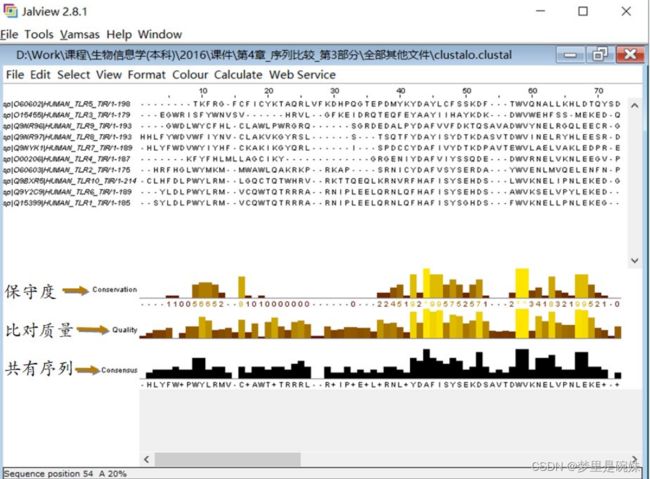
在Colour 菜单下有很多种颜色方案。 能够和保守度这一行柱状图配合的颜色方案是 Percentage Identity。选了这个颜色方案之后, 每一列会根据这一列的保守度用深浅不同的蓝色表示。蓝色越深说明这一列越保守,反之越 不保守。再配合 Colour 菜单下的“By Conservation”参数,可以从弹出的参数设定窗口中设 定保守程度达到百分之多少以上的才给赋予不同的蓝色,阈值以下的都是白色。 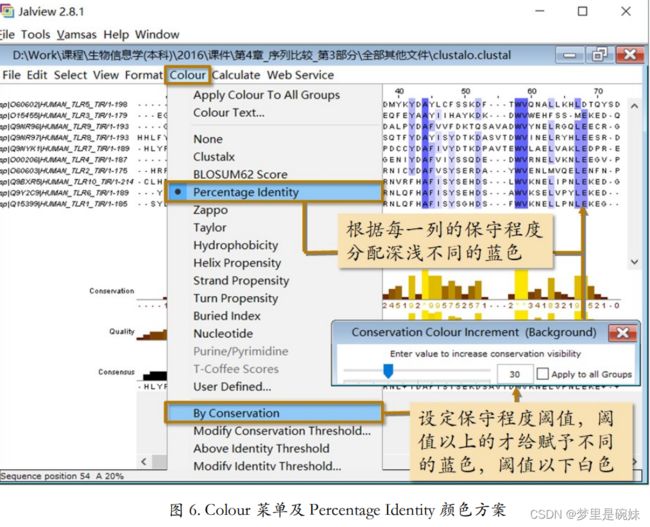
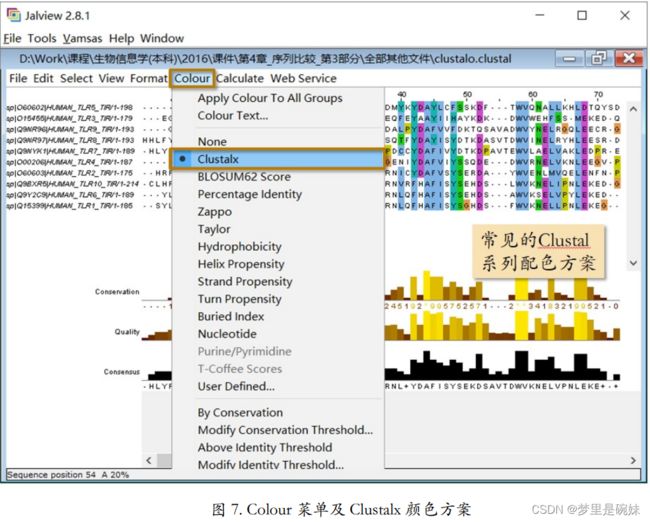
另一个较常用的颜色方案是 Clustal 系列配色方案。这个配色方案和 EMBL 多序列比对 工具做出的结果页面里“Show Colors”之后的颜色方案是基本相同的。
对局部位置进行 手动调整。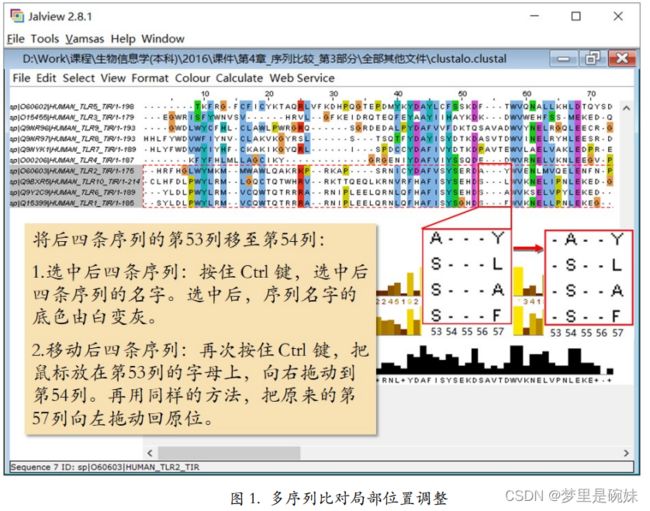
如果想要让多序列比对根据 Jalview 窗口的宽度自动换行,可以在 Format 菜单下勾选“Wrap” 。此外,还可以通过“Font…”窗口对字体格式、大小等进行调整。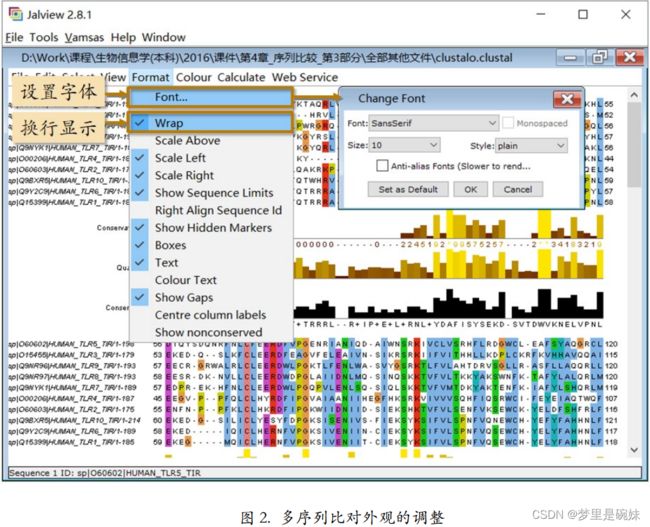
可以按照序列的名字、 两两一致度或其他规则给比对中的序列重新排序以及为选中的两条序列做双序列全局比对。
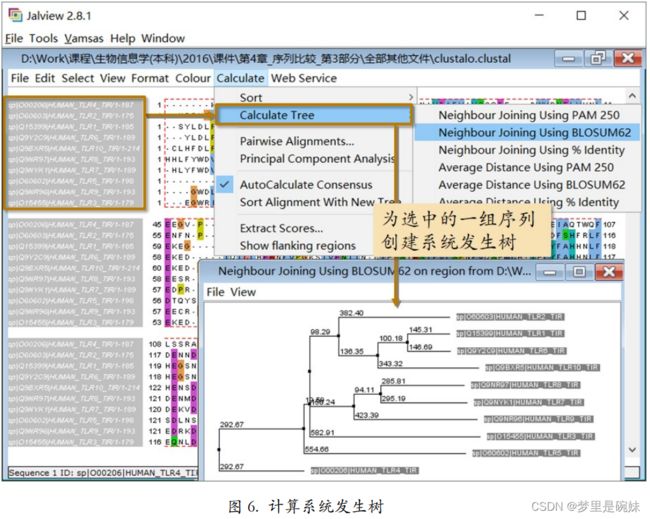
为选中的一组序列计算各种系统发生树。
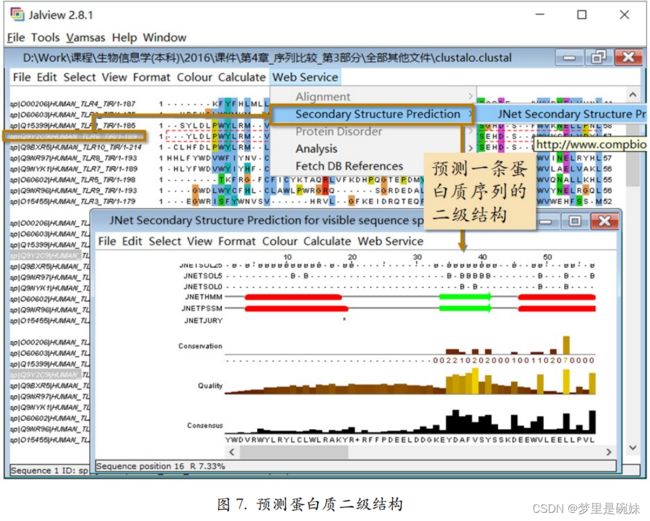
或者用在线软件为某一条序列预测二级结构。
7、寻找保守区域:序列标识图 WebLogo
你究竟想从多序列比对中得到什么?答案是你想要找到序列中重要的位置。说得更专业一点,就是要找到保守区域。也就是序列比对下方的星星点点很多的部分。
:“*”代表这一列残基完全相同;“:”代表这一列残基或者相同或者相似;“.” 代表这一列残基有相似的但也有不相似的;什么都没有代表这一列残基都不相似。
序列标识图就是序列的 logo,它是以图形的方式依次绘出序列比对中各个位置上出现的残基,每个位置上残基的累积可以反 应出该位置上残基的一致性。每个残基对应图形字符的大小与残基在该位置上出现的频率成 正比。 但图形字符的大小并不等于频率百分比,而是经过简单统计计算后转化的结果。图 2 是用一款流行的软件 WebLogo 创建的序列标识图。
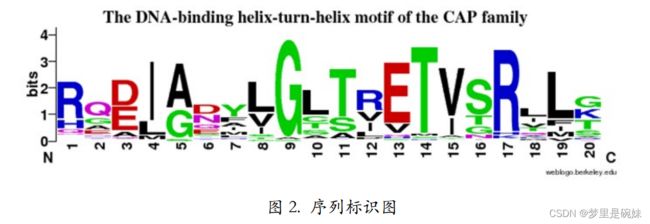
如果某一列非常保守,字母高度就高。反之,如果某一列没有什么特征,各种残基都有出现,杂乱无章,那么就会看到一堆比较矮的字母摞在一起。
再次强调,字母的高度和 它在某一列中出现的频率成正比,但是并不等于频率。试想一下,如果字母高度就是频率的话,那么序列标识图中每个位置上字母摞起来的总高度应该是一样的,都是 100%。但是从图中可以看到,序列标识图上每个位置字母摞起来的总高度是不一样的,这是因为在字母高度的计算过程中涉及了熵值。
某一列中字母出现的情况越混乱,熵值越大,字母越矮。字母出现的情况越有规律,熵值越小,字母越高。所以序列标识图可以很好的展现多序列比对 中每一列的保守程度,即,它们是杂乱无章的,还有有规律可循的。并且把可循的规律图形化的展现出来。这就是我们为什么要给序列打上 logo 的原因。
WebLogo 是一款在线创建序列标识图的软件(http://weblogo.threeplusone.com/)
从图中可以清晰的看到:输入的这些启动子序列上 TATA-Box 的共有特征序列,以及它们出现的位置。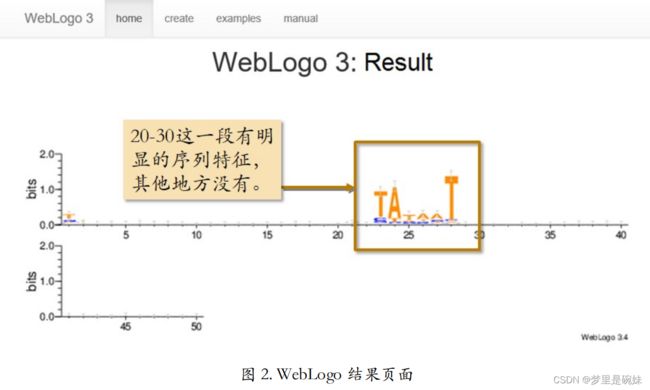
8、寻找保守区域:序列基序 MEME
MEME 是 The MEME Suite 在线软件套装中的一员(http://meme-suite.org/)。MEME 的 使用非常简单,只需要将待分析的序列上传即可。
而且,上传的序列为原始序列, MEME 就是一款可以自动从一组相关的核酸或蛋白质序列中发现序列基序的软件。
不需要提前为它们做多序列比对。你也可以指定返回排名前几的基序。MEME 的等待时间 稍长,大约 10 分钟以上,所以最好留下邮箱。
网页格式的 MEME 结果页面中,给出了找到的排名前三的基序。它们以序列标 识图的形式展现出来。同时还提供这三个基序在每条序列中的大体位置。如果要进一步了解 某个基序,可以点击序列标识图右侧的“More”下面的向下箭头,以查看详细。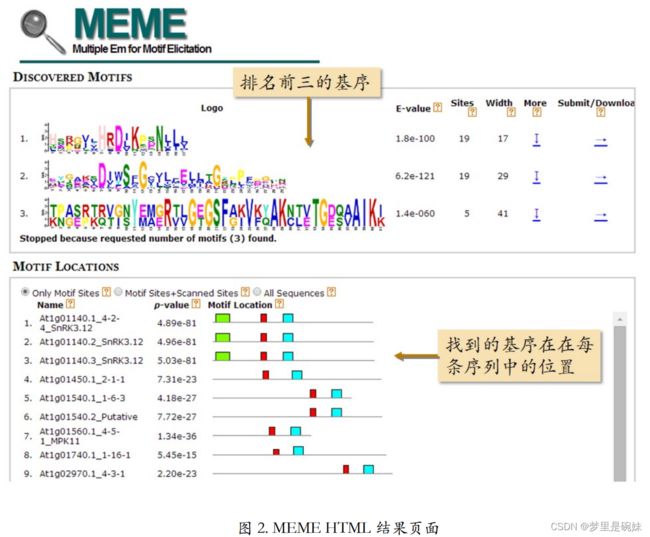
9、寻找保守区域:PRINTS 指纹图谱数据库
目前,科学家已经对现有的蛋白质序列进行了充分的研究,而且早已发现并总结了这些 序列上的重要基序。相关研究成果汇入了 PRINTS 蛋白质序列指纹图谱数据库(http://www. bioinf.manchester.ac.uk/dbbrowser/PRINTS/)。
所谓蛋白质的指纹是指一组保守的序列基序, 用于刻画蛋白质家族的特征。这些基序由多序列比对结果获得,且它们在氨基酸序列水平上是不相邻的,但是在三维结构中可能紧密地结合在一起。PRINTS 数据库存储了目前已发现 的绝大多数蛋白质家族的指纹图谱。对于一个陌生的蛋白质,只要看看它的序列是否符合某个蛋白质家族的图谱就可以对它进行分类并预测它的功能。
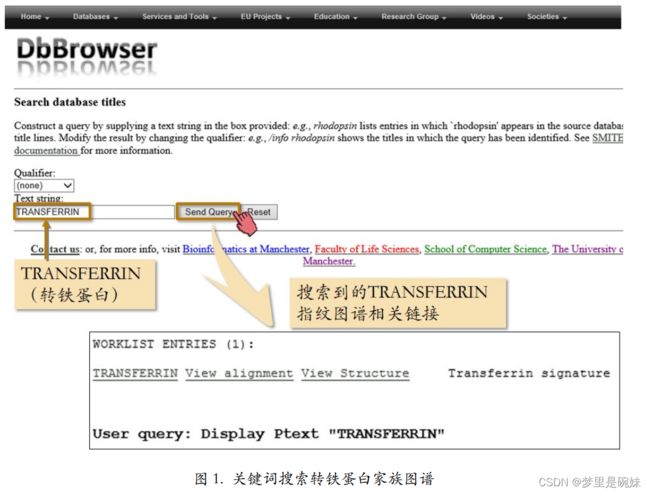
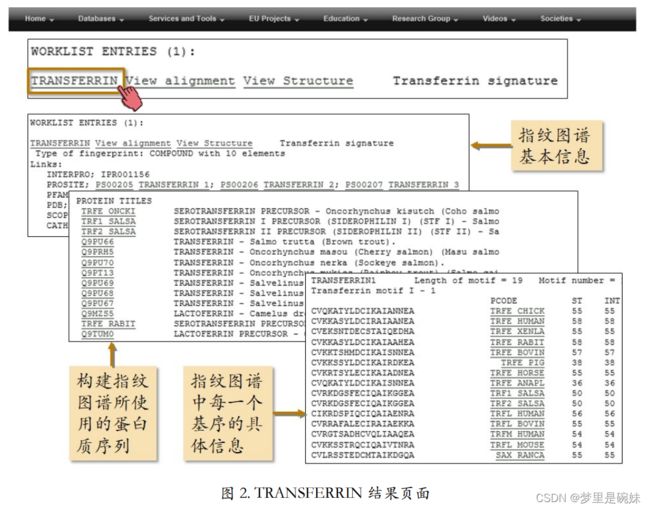
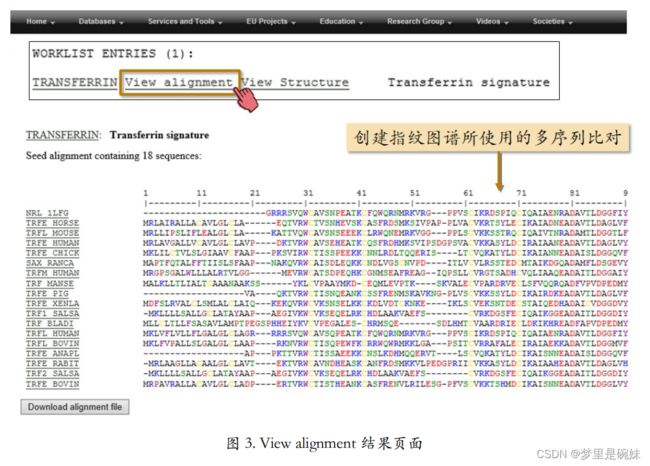
除了浏览某一指纹图谱,PRINTS 还提供指纹匹配服务。也就是搜索某一序列所匹配的 指纹图谱。此功能通过 PRINTS 主页也上的“FPScan”链接实现。注意输入的待搜 索序列只能是“a raw sequence”,也就是纯序列。换言之,FASTA 格式中带大于号的第一行 不能拷贝进输入框。
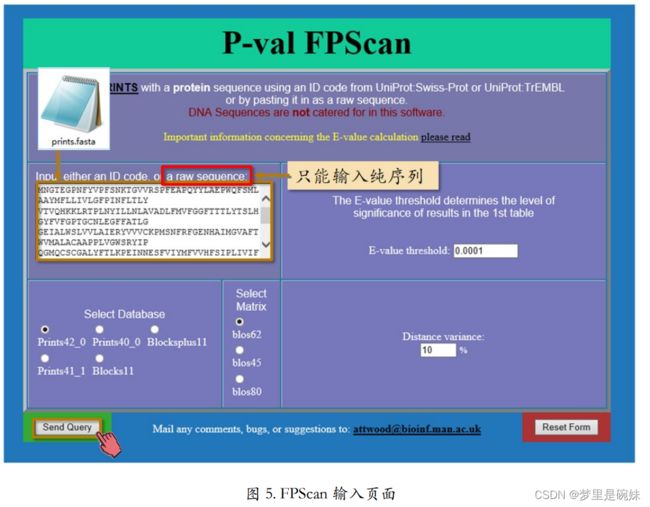
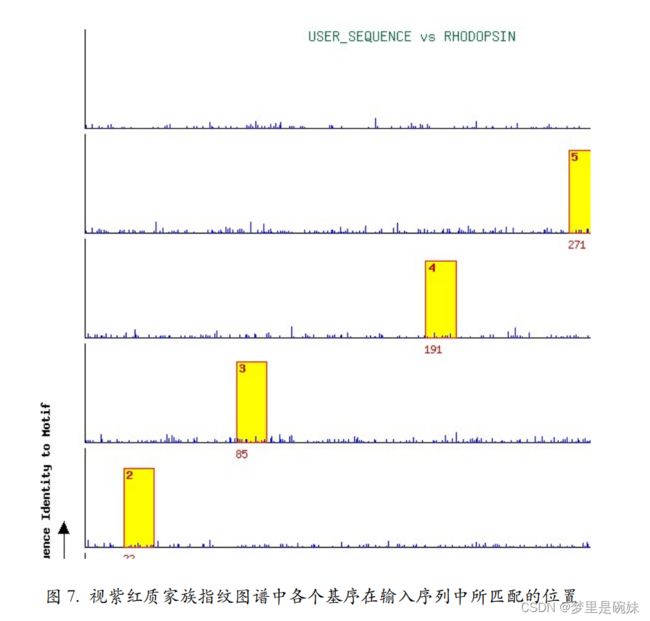
提交后返回的结果页面中,跟输入序列匹配的指纹图谱,根据匹配得分的高低被排列出 来(只列出前十名)。此外,还单独列出了排名前三的指纹图谱。由此可知,得分最高的是视紫红质家族的指纹图谱。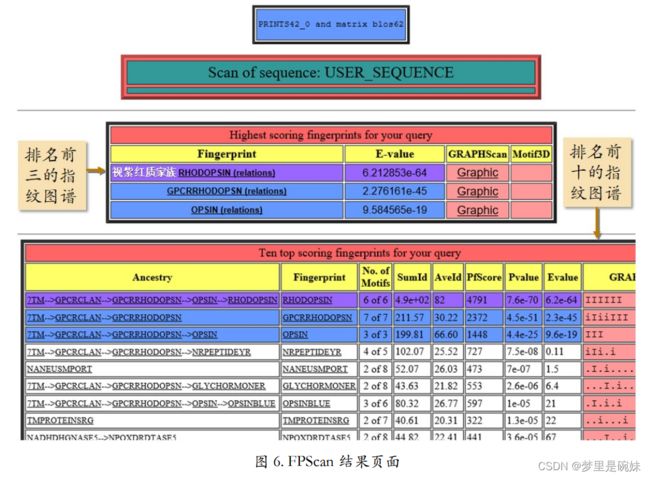
结果页面的下部还提供了视紫红质家族的 6 个基序 在输入序列中所对应的具体序列片段。由此,可以推测,输入序列属于视紫红质家族,并具 有该家族蛋白质的主要功能。