#http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2881355/
一致聚类方法,采用重抽样方法来验证聚类合理性。
library(ALL)
data(ALL)
d=exprs(ALL)
d[1:5,1:5]

#对上面这个芯片表达数据我们一般会简单的进行normalization (本次采用中位数中心化),然后取在各个样品差异很大的那些gene或者探针的数据来进行聚类分析
mads=apply(d,1,mad)# mad(x) 绝对中位数差 按行(1)取d数据的中位数
d=d[rev(order(mads))[1:5000],]
#去除前5000个数据
d = sweep(d,1, apply(d,1,median,na.rm=T))
#按行减去中位数,r语言中使用sweep(x, MARGIN, STATS, FUN="-", ...) 对矩阵进行运算。MARGIN为1,表示行的方向上进行运算,
#为2表示列的方向上运算。STATS是运算的参数。FUN为运算函数,默认是减法。
library(ConsensusClusterPlus)
title=tempdir()
results = ConsensusClusterPlus(d,maxK=6,reps=50,pItem=0.8,pFeature=1,
clusterAlg="hc",distance="pearson",seed=1262118388.71279,plot="png")
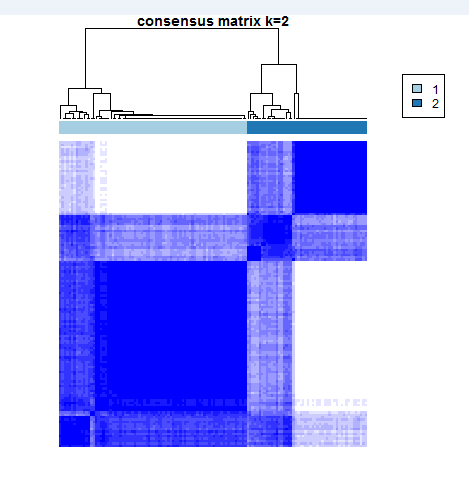
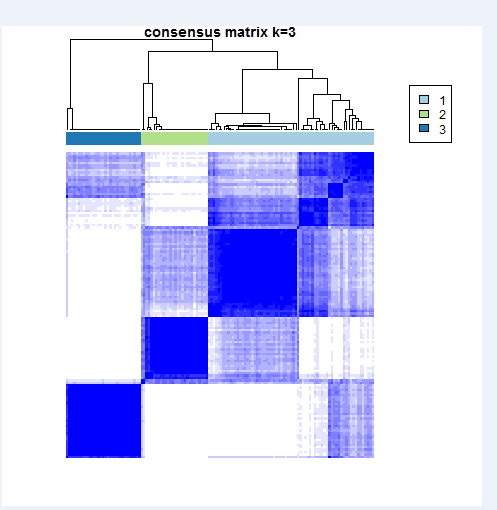
#聚类数目K=2,3,4,·····6,采用重抽样方案对样本的80%抽样,经过多次采样,找到稳定可靠的亚组分类。
然后利用这些有类标签的样本来寻找可以将样本分类的标签基因。可以利用PAM方法找寻标签基因。
#results[[2]] is theresults result of k=2
results[[4]][["consensusMatrix"]][1:5,1:5]
results[[2]][["consensusTree"]]
results[[2]][["consensusClass"]][1:5]
icl = calcICL(results,title=title,plot="png")
icl[["clusterConsensus"]]
icl[["itemConsensus"]][1:5,]
生成的图片类似: