Accurate prediction of protein contact maps by coupling residual two-dimensional bidirectional long
论文题目:Accurate prediction of protein contact maps by coupling residual two-dimensional bidirectional long short-term memory with convolutional neural networks
下载链接:https://academic.oup.com/bioinformatics/article/34/23/4039/5040307 or https://doi.org/10.1093/bioinformatics/bty481
题目翻译:基于残差二维双向长短期记忆和卷积神经网络的蛋白质接触图精确预测
Abstract
Motivation:Accurate prediction of a protein contact map depends greatly on capturing as much contextual information as possible from surrounding residues for a target residue pair. Recently, ultra-deep residual convolutional networks were found to be state-of-the-art in the latest Critical Assessment of Structure Prediction techniques (CASP12) for protein contact map prediction by attempting to provide a protein-wide context at each residue pair. Recurrent neural networks have seen great success in recent protein residue classification problems due to their ability to propagate information through long protein sequences, especially Long Short-Term Memory (LSTM) cells. Here, we propose a novel protein contact map prediction method by stacking residual convolutional networks with two-dimensional residual bidirectional recurrent LSTM networks, and using both one-dimensional sequence-based and two-dimensional evolutionary coupling-based information.
动机:蛋白质接触图的准确预测很大程度上取决于从目标残基对的周围残基中获取尽可能多的上下文信息。最近,超深残差卷积网络在CASP12中被认为是最新的蛋白质接触图预测技术,它试图在每个残基对上提供一个蛋白质范围的上下文。由于递归神经网络能够通过长的蛋白质序列传递信息,特别是长短时记忆(LSTM)细胞,因此在蛋白质残基分类问题中取得了巨大的成功。在这里,我们提出了一种新的蛋白质接触图预测方法,该方法将残差卷积网络与二维残差双向递归LSTM网络叠加,同时利用一维序列和二维进化耦合信息。
Results:We show that the proposed method achieves a robust performance over validation and independent test sets with the Area Under the receiver operating characteristic Curve (AUC) > 0.95 in all tests. When compared to several state-of-the-art methods for independent testing of 228 proteins, the method yields an AUC value of 0.958, whereas the next-best method obtains an AUC of 0.909. More importantly, the improvement is over contacts at all sequence-position separations. Specifically, a 8.95%, 5.65% and 2.84% increase in precision were observed for the top L∕10 predictions over the next best for short, medium and long-range contacts, respectively. This confirms the usefulness of ResNets to congregate the short-range relations and 2D-BRLSTM to propagate the long-range dependencies throughout the entire protein contact map ‘image’.
结果表明,在所有测试中,该方法在验证集和独立测试集上都具有良好的鲁棒性,ROC曲线下方的面积(AUC)> 0.95。在独立测试集(228种蛋白质)上与几种最新方法相比,该方法的AUC值为0.958,而次优方法的AUC值为0.909。更重要的是,改进是在所有序列位置分离的接触。更重要的是,在所有序列位置分离的接触上都有所改善。具体地说,对于短距离、中距离和长距离接触,top L/10预测的精度分别提高了8.95%、5.65%和2.84%。这证实了传播短距离关系的ResNets和传播长程依赖的2D-BRLSTM在整个蛋白质接触图图像中非常有用。
【注:AUC(Area Under Curve)被定义为ROC曲线下与坐标轴围成的面积,显然这个面积的数值不会大于1。又由于ROC曲线一般都处于y=x这条直线的上方,所以AUC的取值范围在0.5和1之间。AUC越接近1.0,检测方法真实性越高;等于0.5时,则真实性最低,无应用价值。】
Availability and implementation:SPOT-Contact server url: http://sparks-lab.org/jack/server/SPOT-Contact/.
Supplementary information:Supplementary data are available at Bioinformatics online.
1 Introduction
Proteins are one of the most biologically important macromolecules with a wide variety of functions. Because the functions of most proteins rely on their uniquely-folded three-dimensional structures, determining protein structures is of great importance to understand functional mechanisms. Due to the high cost and low efficiency of experimental techniques, ab initio prediction of protein structures by computational methods have been actively pursued in the past 50 years, but are still yet to be solved. To simplify the problem, breaking the structure-prediction problem into more feasible sub-problems has been the forefront of bioinformatics studies for decades.
蛋白质是最重要的生物大分子之一,具有多种功能。由于大多数蛋白质的功能依赖于其独特的折叠三维结构,因此确定蛋白质结构对于理解其功能机制具有重要意义。由于实验技术的高成本和低效率,利用计算方法进行蛋白质结构的从头算预测是近50年来研究的热点,但仍有待解决。为了简化问题,将结构预测问题分解为更可行的子问题一直是几十年来生物信息学研究的前沿。
One such sub-problem is the prediction of residue-residue contacts. By analyzing whether or not a residue pair in a protein sequence is in contact (i.e. close in 3D space), we are able to form a protein contact map, which provides key structural restraints towards the modeling of a protein’s three-dimensional structure. The current methods for predicting protein contact maps can be sorted into two distinct groups: Evolutionary Coupling Analysis (ECA) and machine learning techniques.
一个这样的子问题是残基接触的预测。通过分析蛋白质序列中的残基对是否接触(即在三维空间中接近),我们能够形成蛋白质接触图,这为蛋白质三维结构的建模提供了关键的结构约束。目前预测蛋白质接触图的方法可分为两大类:进化耦合分析(ECA)和机器学习技术。
ECA methods utilize Multiple Sequence Alignments (MSAs; Göbel et al., 1994) to identify correlation in changing (co-evolving) residue pairs, using the belief that residues in close proximity mutate in sync with the evolutionary functional and structural requirements of a protein. These methods have benefited from the explosion of available protein sequences in the past decade, as these methods perform particularly well when a target protein sequence has a high number of homologues in a protein database (Ovchinnikov et al., 2017). Popular ECA methods include: CCMPred (Seemayer et al., 2014), FreeContact (Kaján et al., 2014), GREMLIN (Kamisetty et al., 2013), PlmDCA (Ekeberg et al., 2013) and PSICOV (Jones et al., 2012). While these methods are useful for predicting long-range contacts in proteins with a high number of sequence homologues, their accuracy is poor if the number of homologues is low (Wang and Xu, 2013).
ECA方法利用多重序列比对来识别变化(共同进化)残基对中的相关性,使用的信念是近距离的残基突变与蛋白质的进化功能和结构要求同步。这些方法得益于近十年来可用蛋白序列的激增,因为当目标蛋白序列在蛋白质数据库中具有大量同源序列时,这些方法表现尤其出色。常用的ECA方法包括:CCMPred , FreeContact , GREMLIN , PlmDCA 和 PSICOV。虽然这些方法对于预测具有大量同源序列的蛋白质中的长程接触是有用的,但是如果同源序列的数目较少,它们的准确性就很差。
The other, increasingly accurate methods are based on machine learning techniques. These methods have seen success due to their ability to learn underlying relationships present in sequence-based features given a set of labelled data. They have been found especially effective when predicting on proteins with few homologues. Early machine learning papers utilized Support Vector Machines (SVM; Vapnik, 1998) due to their ability to model complex relationships despite a lack of processing power and extensive data, such as SVMCon (Cheng and Baldi, 2007), SVMSEQ (Wu and Zhang, 2008) and the recently-released R2C (Yang et al., 2016). Other papers have found success in exploiting the ever-increasing amount of available training data by the application of Deep artificial Neural Networks (DNN’s; Rumelhart et al., 1985) in various forms, such as two-dimensional (2D) Recursive NNs (Rec-NN’s; Baldi and Pollastri, 2003) and Deep Belief Network’s (Hinton et al., 2006). Such predictors include Betacon (Cheng and Baldi, 2005), CMAPPro (Di Lena et al., 2012), DeepConPred (Xiong et al., 2017) and NNCon (Tegge et al., 2009).
另一种越来越精确的方法是基于机器学习技术的。这些方法之所以成功,是因为它们能够在给定一组标记数据的情况下,学习序列特征中的潜在关系。在预测同源物较少的蛋白质时,它们被发现特别有效。早期的机器学习论文使用支持向量机,因为它们能够在缺乏处理能力和大量数据的情况下建模复杂的关系,例如SVMCon、SVMSEQ以及最近发布的R2C。其他论文发现,通过应用各种形式的深层人工神经网络(DNN),如二维(2D)递归神经网络(Rec-NN)和深度信念网络,成功地利用了不断增加的可用训练数据量。这些预测器包括Betacon、CMAPPro、DeepConPred和NNCon。
Complementary methods can be combined in the form of metapredictors, where a single network combines the outputs of several other classifiers. Examples of this architecture are MetaPSICOV (Jones et al., 2015) and NeBCON (He et al., 2017). MetaPSICOV received the best prediction results in a recent review by Wuyun et al. (2016).
补充方法可以以各种元预测器的形式组合,其中一个网络结合了其他几个分类器的输出。这种架构的例子有MetaPSICOV和NeBCON。MetaPSICOV在Wuyun等人最近的一次评估中获得了最好的预测结果。
The recently-released RaptorX-Contact (Wang et al., 2017) and DNCON2 (Adhikari et al., 2017) predictors are the first approaches to attempt the incorporation of the entire protein ‘image’ as context for prediction. The architecture utilized in these models are Convolutional NN’s (CNN; LeCun et al., 1989), with RaptorX-Contact utilizing a Residual CNN, or ResNet (He et al., 2016a). ResNets achieve identity mappings between several layers by employing shortcut connection between the output of a previous layer and the current output. This allows these models to have ultra-deep architectures due to their ease of propagating the error gradient through many layers, and have been shown to benefit from having over 100 convolutional layers. RaptorX-Contact is currently the state-of-the-art predictor for the latest Critical Assessment of Structure in Proteins (CASP) round, demonstrating that access to the entire protein as context is greatly beneficial to learning contact maps (Schaarschmidt et al., 2018; Wang et al., 2018). It also is one of the techniques to benefit from combining sequence-derived features with the information from evolution coupling (Betancourt and Thirumalai, 1999; Miyazawa and Jernigan, 1985). DNCON2, on the other hand, can be split into two sections. The first uses a set of intermediate CNNs to predict contact maps at several distances (from 6–10 Å). It then combines these separate predictions in a secondary CNN to provide the final contact map at 8 Å.
最近发布的 RaptorX-Contact和DNCON2预测器是首次尝试将整个蛋白质“图像”作为上下文进行预测的方法。这些模型中使用的结构是卷积神经网络(CNN),RaptorX-Contact利用残差CNN或ResNet。ResNet通过在前一层的输出和当前输出之间使用shortcut connection来实现多个层之间的身份映射。这使得这些模型具有超深的体系结构,因为它们易于通过许多层传播误差梯度,并且已经被证明拥有100多个卷积层非常有益。RaptorX-Contact是目前最新的蛋白质结构关键性评估(CASP)的最新预测器,表明获取整个蛋白质作为上下文对学习接触图非常有利。它也是将序列特征和进化耦合信息相结合的技术之一。另一方面,DNCON2可分为两部分。第一种方法使用一组中间CNN来预测不同距离(6–10 埃)的接触图 。然后将这些单独的预测合并到第二个CNN中,以提供8 埃 的最终接触图。
The neural network architecture utilized in this paper was inspired by RaptorX-Contact and the Multi-Dimensional Recurrent Neural Network (MD-RNN) in Graves et al. (2007), in which stacked 2D RNNs were proven able to progress information throughout entire 2D images. The advantages here were that the model was able to generalize along all input spatio-temporal dimensions, making the model robust to distortions in any mixture of the input dimensions, with the architecture performing particularly well on warped data in comparison to CNNs. The MD-RNN was simplified in ReNets (not to be confused with ResNets), where the x and y dimensions’ forward and backward RNNs were separated between consecutive layers (Visin et al., 2015). Min et al. (2017) described the MD-RNN as an emerging architecture in bioinformatics in a recent review.
本文中使用的神经网络体系结构受RaptorX-Contact和Graves等人中的多维递归神经网络(MD-RNN)的启发,其中堆叠的2D-RNN能够在整个2D图像中传递信息。这里的优点是,该模型能够沿所有输入时空维度进行泛化,使得模型对输入维度的任何混合情况都具有鲁棒性,与cnn相比,该体系结构在扭曲数据上的表现尤其出色。在ReNets中,MD-RNN被简化(不要与 ReNets混淆),其中x和y维的前向和后向RNN在连续层之间分开。Min等人在最近的一篇综述中将MD-RNN描述为生物信息学的一个新兴架构。
In combination with RNNs, Long Short-Term Memory (LSTM) cells (Hochreiter and Schmidhuber, 1997) are commonly used to model long-range context which is vital to modeling complex relationships between non-local datapoints. Bidirectional LSTM networks (Schuster and Paliwal, 1997) have already seen success in bioinformatics applications, where their effective propagation of deep residue structural interdependencies have provided state-of-the-art results in protein secondary structure prediction (Heffernan et al., 2017) and protein disorder prediction (Hanson et al., 2017), the latter of which demonstrating that LSTM cells are able to accurately predict sparse data, an aspect shared between protein disorder and contact map prediction.
与RNN相结合,长短期记忆(LSTM)单元通常用于对长程上下文进行建模,这对于建模非局部数据点之间的复杂关系至关重要。双向LSTM网络已经在生物信息学应用中取得了成功,其中,它们对深残差结构相互依赖性的有效传播在蛋白质二级结构预测和蛋白质乱序预测提供了最新成果,后者证明LSTM细胞能够准确预测稀疏数据,这是蛋白质乱序和接触图预测的共同点。
In this paper, we aim to capture these deep, underlying relationships between non-local residue pairs in both spatial dimensions for protein contact map prediction by the use of an ultra-deep hybrid network, consisting of a ResNet coupled with 2D Bidirectional-ResLSTMs (2D-BRLSTM). Using this technique, the proposed method, called SPOT-Contact (Sequence-based Prediction Online Tools for Contact map prediction), is able to capture contextual information from the whole protein ‘image’ at each layer. SPOT-contact has been found to be highly accurate for predicting contacts at all sequence-position separations, significantly outperforming all methods compared.
在这篇论文中,我们的目的是利用一个超深的混合网络,包括一个ResNet和2D Bidirectional-ResLSTMs (2D-BRLSTM)组成的超深混合网络,在两个空间维度上捕捉非局部残基对之间的深层次关系,以预测蛋白质接触图。利用这项技术,所提出的方法称为SPOT-Contact(基于序列的预测在线工具,用于接触图预测),能够从每一层的整个蛋白质图像中获取上下文信息。SPOT-contact被发现在所有序列位置分离处预测接触都是非常准确的,显著优于所有的方法。
2 The machine learning approach
2.1 Ensemble of two-dimensional bidirectional recurrent neural networks and ResNets
Our approach to the problem is built up of an ensemble of models, all based on slight variations of the network architecture shown in Figure 1. The base model can be broken down into four separate segments: input preparation, ResNet, 2D-BRLSTM and Fully-Connected (FC).
我们解决这个问题的方法是由一组模型组成的,所有模型都基于图1所示的网络体系结构的细微变化。基本模型可以分为四个独立的部分:输入准备、ResNet、2D-BRLSTM和全连接(FC)。
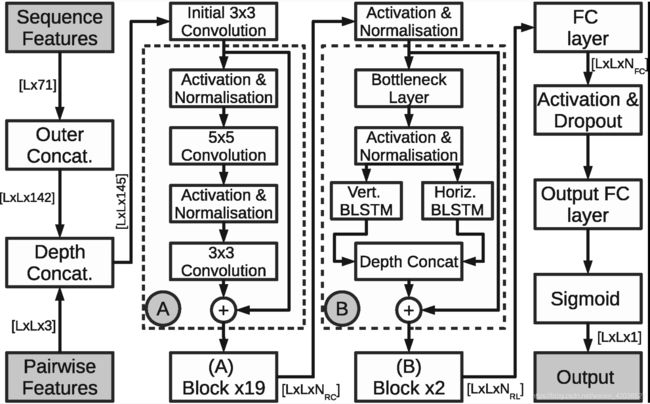
Figure 1:The network layout of the SPOT-Contact. The ResNet (Residual Convolutional Neural Network) and 2D-BRLSTM (2-Dimensional Bidirectional Residual LSTM Network) functions are provided in boxes ‘A’ and ‘B’, respectively. NRC, NRL, and NFC are the depth of the CNN filters in the ResNet block, four times the depth of the LSTM layers in the 2D-BRLSTM block and the depth of the FC layer, respectively, and L is the length of the input protein. Depth concatenation means concatenation along the last dimension
The data preparation segment involves the transformation of our sequence-based one-dimensional features into a two-dimensional representation. This is achieved through the outer concatenation function, as described in Wang et al. (2017). However, rather than concatenating the features of residues i,j and (i+j)/2 at position (i, j), we omit the concatenation of the midpoint residue. The product of the concatenation stage is then depth concatenated (i.e. concatenated along the feature axis) with the two-dimensional features.
数据准备部分包括将基于序列的一维特征转换为二维表示。这是通过外部连接函数实现的,如Wang等人所述。然而,我们没有在位置(i,j)连接残基i,j和(i+j)/2的特征,而是省略了中点残基的连接。然后,串联阶段的产物与二维特征进行深度连接(即沿特征轴连接)。
The ResNets used in our model utilize the pre-activation order of operations as proposed in He et al. (2016b). As such, an initial convolutional layer was placed before the first residual block. The residual block architecture is shown in the ResNet Block section of Figure 1. The output of the entire ResNet was also activated and normalized prior to the 2D-BRLSTM section. The size of the convolutions alternated between a kernel size of 3 x 3 and 5 x 5, both with 60 filters and had a Exponential Linear Unit (ELU) activation (Clevert et al., 2015). ELU activations have been seen to be more effective than the standard ReLU activation function in learning for ResNets (Shah et al., 2016).
我们模型中使用的ResNets利用了He等人提出的预激活操作顺序。因此,在第一个残差块之前放置初始卷积层。剩余块架构如图1的ResNet块部分所示。整个ResNet的输出在2D-BRLSTM部分之前也被激活和规范化。具有5个单位的线性卷积和5个单位的线性卷积。卷积的大小在核大小为3 x 3和5 x 5之间交替变化,都有60个滤波器,并且具有指数线性单位(ELU)激活。在ResNets学习中,ELU激活比标准ReLU激活函数更有效。
The 2D-RNNs used in this model differ to Graves et al. (2007) by the fact that each of the four directions’ RNNs in each layer is completely independent of all of the other directions’ due to limitations in the coding environment, as is similar to Visin et al. (2015). Although each directions’ outputs are calculated separately, they are concatenated at the output of each layer to provide information from the entire 2D image plane to the next layer. However, He et al. (2016a) discussed that the identity mapping function is less effective at error propagation when the residual connection is connected over only a single layer’s activation. Therefore, we add a bottleneck layer before the LSTM layers to increase the depth of the residual connection. This also has the added benefit of reducing the parameter count of the model. The bottleneck and LSTM layers form our BRLSTM blocks (Kim et al., 2017) as shown in Figure 1. This bottleneck connection is established by a 1 × 1 convolution with an ELU activation.
该模型中使用的2D-RNN与Graves等人(2007)的不同之处在于,由于编码环境的限制,四个方向中的每一个“每层RNN”都完全独立于所有其他方向,这与Visin等人(2015)类似。虽然每个方向的输出是分开计算的,但是它们在每一层的输出处被连接起来,以提供从整个2D图像平面到下一层的信息。然而,He等人(2016a)讨论了当残差连接仅通过单个层的激活连接时,身份映射函数在错误传播方面的效率较低。因此,我们在LSTM层之前添加 bottleneck层,以增加残差连接的深度。这还有一个额外的好处,即减少模型的参数计数。bottleneck层和LSTM层构成了我们的BRLSTM区块,如图1所示。这种bottleneck连接是通过1 × 1卷积和ELU激活建立的。
The default 2D-BRLSTM layer’s LSTM cells consist of 200 one-cell memory blocks, culminating in 800 inputs at the succeeding layer. The FC layers consist of 400 nodes plus a bias node with an ELU activation, except at the output layer which has a single output neuron and a sigmoid activation to convert the output into a likelihood of a residue pair being in contact. The network layout from Figure 1 is only changed when omitting the bottleneck layer, and when the 2D-BRLSTM is placed first in the network (see the model variants listed in Supplementary Table S1). All of the parameters discussed above were chosen after thorough experimentation, during which this architecture was found to obtain consistently high accuracies through short, medium and long distance contacts on a validation set, yet fitting into the strict computational memory constraints of a 2D-BRLSTM.
默认的2D-BRLSTM层的LSTM单元由200个单元内存块组成,最后在下一层输入800个。FC层由400个节点加上一个ELU激活的偏置节点组成,除了输出层有一个输出神经元和一个sigmoid激活将输出转换为残基对接触的可能性。图1中的网络布局仅在省略bottleneck层和2D-BRLSTM首先放置在网络中时更改(参见补充表S1中列出的模型变量)。上面讨论的所有参数都是经过深入的实验后选择的,在此过程中,我们发现该体系结构通过验证集上的短、中、长距离接触获得一致的高精度,同时也符合2D-BRLSTM严格的计算内存限制。
Each model was trained with the ADAM optimization algorithm (Kingma and Ba, 2014), which has been shown to converge more quickly than the traditionally-used Stochastic Gradient Descent with the additional benefit of having standard hyperparameters which require little to no tuning. Regularization was applied to the network through the use of layer normalization (Lei Ba et al., 2016) at each normalization block in Figure 1, and a 50% dropout rate at the output of the FC layer during training (Srivastava et al., 2014).
每个模型均采用ADAM优化算法进行训练,该算法比传统的随机梯度下降法收敛更快,同时还具有标准超参数(几乎不需要调整)的额外好处。通过在图1中的每个标准化块使用层标准化,以及训练期间FC层输出的50%的dropout率,将正则化应用到网络中。
Using an ensemble predictor allowed us to minimize the effects of generalizations on the data. Because the tuned weights of a neural network learn slightly different representations of the data (due to various weight initializations and training data feeding), these lead to various errors at the output which are dependent on the generalizations made. Assuming that the correct outputs should be more common between the individual predictors, the collective decision between all of the predictors should be less likely to contain the errors pertaining to an individual predictor’s generalization (Hansen and Salamon, 1990). All six models (base network, base without bottleneck, base without FC, 2D-BRLSTM prior to ResNet in the base model and the 2D-BRLSTM only model) used in SPOT-Contact are shown along with their network parameters in Supplementary Table S1. The results of SPOT-Contact are provided by the mean of all six networks’ outputs. An ensemble of models was also utilized in RaptorX-Contact.
使用集成预测器可以使我们最小化泛化对数据的影响。由于调整后的神经网络权值学习的数据表示略有不同(由于不同的权值初始化和训练数据喂送),这些会导致输出中的各种错误,这些错误取决于所做的泛化。假设正确的输出应该在各个预测值之间更常见,那么所有预测值之间的集体决策应该不太可能包含与单个预测值泛化相关的错误。用于SPOT-Contact的所有六个模型(基本网络、base without bottleneck、base without FC、 2D-BRLSTM prior to ResNet in the base model and the 2D-BRLSTM only model)及其网络参数见补充表S1。SPOT-Contact 的结果由所有六个网络输出的平均值提供。在RaptorX-Contact中也使用了一组模型。
Training of the model was executed in the framework of Google’s Tensorflow library (v1.4; Abadi et al., 2016), enabling us to accelerate the training of the model by training the model on an Nvidia GTX TITAN X Graphics Processing Unit (GPU). Oh and Jung (2004) showed that the use of a GPU in neural network training can speed up the total training time by up to a factor ≈20. Total training time was mostly influenced by the depth of the 2D-BRLSTM layers, with each network taking roughly 50 h for 15 epochs over our whole training set. The size of the 2D-BRLSTM layers dictated the memory consumption during training. Thus, the model hyperparameters, such as LSTM cell size, were chosen as a compromise between the training time, memory usage and performance of the model. Deeper and larger architectures were tested by spreading the model over multiple GPU’s, but this was not found to improve performance significantly.
模型的训练是在谷歌Tensorflow库的框架下进行的,使我们能够通过在Nvidia GTX TITAN X图形处理单元(GPU)上训练模型来加快模型的训练。Oh和Jung(2004)指出,在神经网络训练中使用GPU可以将总训练时间提高一倍≈20。总的训练时间主要受2D-BRLSTM层深度的影响,在我们的整个训练集中,每个网络在15个周期内大约需要50 h。在BRLSTM-2D训练过程中决定了内存消耗的大小。因此,模型的超参数(如LSTM单元大小)被选作训练时间、内存使用和模型性能之间的折衷。通过将模型扩展到多个GPU上,对更深更大的体系结构进行了测试,但没有发现这能显著提高性能。
2.2 Input features
The inputs to our model included both one-dimensional (i.e. along the primary sequence) and two-dimensional features (i.e. pairwise, or per residue pair). One-dimensional features consisted of the Position-Specific Scoring Matrix (PSSM) profile, the HMM profile from HHblits (Remmert et al., 2012) and several predicted structural probabilities from SPIDER3 (Heffernan et al., 2017). The PSSM profile was generated by three iterations of PSI-BLAST (Altschul et al., 1997) against the UniRef90 sequence database updated in April 2018. The HMM profile was generated by hhblits v3.0.3 with default parameters by searching the UniClust30 profile HMM database updated in October 2017 (Mirdita et al., 2016). The predicted values obtained from SPIDER3 were: 1 relative solvent-Accessible Surface Area, 2 Half-Sphere Exposures based on the Cα atom, the 8 sine/cosines of the backbone torsion angles (theta, tau, phi and psi) and the three probabilities of the predicted secondary structure. Finally, we also employed a set of seven physicochemical properties provided by Meiler et al. (2001). This gives a total of 71 1D features for the initial section of the network.
我们的模型的输入既包括一维特征(即沿着主序列)和二维特征(即成对的,或每个残基对)。一维特征包括位置特异性评分矩阵(PSSM)profile、HHblits的HMM profile和SPIDER3的几个预测结构概率。PSSM profile 是根据2018年4月更新的UniRef90序列数据库,通过三次PSI-BLAST迭代生成的。通过搜索2017年10月更新的UniClust30 profile HMM数据库,hhblits v3.0.3使用默认参数生成HMM配置文件。由SPIDER3得到的预测值为:1个相对溶剂可及表面积,2个基于Cα原子的半球曝光量,8个主链扭转角(θ、tau、phi和psi)的正余弦和预测二级结构的三个概率。最后,我们还使用了Meiler等人提供的七种物理化学性质。这为网络的初始部分提供了总共71个1D特性。
The 2D features consist of the output from CMMPred (Seemayer et al., 2014), and 2 outputs (mutual and direct-coupling information) from DCA (Morcos et al., 2011), resulting in three pairwise features for concatenation with the output of the first section of the network. The data was standardized to have zero mean and unit variance (according to the training data) before being input into the network.
2D特征包括来自CMMPred的输出和DCA的2个输出(相互和直接耦合信息),产生了三个成对特征,用于与网络第一部分的输出连接。在输入网络之前,将数据标准化为零均值和单位方差(根据训练数据)。
2.3 Datasets
We downloaded 30% non-redundant sequences with resolution <2.5 Å and R-factor < 1.0 from the cullpdb website, which contained 14 541 chains on February 2017. After removing obsolete sequences, sequences containing less than 30 residues and proteins greater than 25% sequence similarity according to BlastClust (Altschul et al., 1997), we kept 12 450 chains. In order for a good comparison with other methods, we have kept all 1250 chains deposited after June 2015 as our independent test set (Test) and the remaining 11 200 as training set. We produced a difficult (‘hard’) subset of Test (Test-Hard), by removing any proteins in Test that have a PSI-Blast E-value of 0.1 or less to any proteins in the training set (i.e. further removing any proteins with potential homologous relations to the training set). This ‘hard’ dataset contains 280 chains.
我们在2017年2月从cullpdb网站下载了分辨率<2.5 Ρ和R-factor<1.0的30%非冗余序列,该网站包含14541条链。根据BlastClust,去除过时序列、含有少于30个残基的序列和大于25%序列相似性的蛋白质后,我们保留了12450条链。为了与其他方法进行比较,我们保留了2015年6月之后存放的所有1250条链作为我们的独立测试集(Test),剩下的11200条链作为训练集。我们产生了一个不同的(“hard”)测试子集(Test-Hard),通过去除训练集中PSI-Blast E值为0.1或更低的任意蛋白质(即进一步去除与训练集有潜在同源关系的任何蛋白质)。这个‘hard’数据集包含280条链。
Due to limitations of the coding environment imposed by the large memory usage by the 2D-BRLSTM model, training and testing input proteins are limited to proteins of length ≤ 300 and ≤ 700 residues, respectively. While this restriction excludes many proteins, it still incorporates upwards of 90% of single domain sequences for testing with our model (Islam et al., 1995). This restriction left us with 7557 training proteins, and a validation, Test and Test-Hard sets of 983, 1213 and 277 proteins, respectively. We also obtained 22 template-free modeling (TFM) CASP12 targets as an additional test set. Their sequence similarity to our training set is also <25% according to BlastClust. All of these datasets can be obtained from www.sparks-lab.org.
由于2D-BRLSTM模型大内存使用所带来的编码环境的限制,输入蛋白的训练和测试长度分别限于≤300和≤700个残基。虽然这一限制排除了许多蛋白质,但它仍然包含了90%以上的单域序列,以便用我们的模型进行测试。这一限制给我们留下了7557个训练蛋白,以及983、1213和277个蛋白质的验证、测试和Test-Hard 。我们还获得了22个无模板的CASP12靶点作为附加测试集。根据BlastClust,它们与我们训练集的序列相似性也<25%。所有这些数据集都可以从 www.sparks-lab.org获得。
To gauge the performance increase by training on new sequence profiles, we also trained an exact replica of SPOT-Contact using the UniRef and UniProt datasets from March 2017 and February 2016, respectively. This model, named SPOT-Contact-2016 will serve as a baseline to compare the other predictors to, to illustrate that the performance improvement reported here is not solely caused by a simple update of sequence libraries.
为了通过新序列profiles的训练来衡量性能的提高,我们还分别使用2017年3月和2016年2月的UniRef和UniProt数据集训练了一个精确的SPOT-Contact副本。这个名为SPOT-Contact-2016的模型将作为比较其他预测器的基线,以说明这里报告的性能改进不仅仅是由序列库的简单更新引起的。
2.4 Performance evaluation
Protein residues in these experiments are considered to be in contact when the inter-residue distance between the two Cβ atoms is ≤ 8.0 Å, following the standard CASP definition (Ezkurdia et al., 2009). For a protein of length L, we first separate it into short ([Math Processing Error]12>|i−j|≥6), medium ([Math Processing Error]24>|i−j|≥12) and long ([Math Processing Error]|i−j|≥24) sequence-position-separated residues. From these groups, we take the top L/k highest-ranked predictions (where [Math Processing Error]k∈{10,5,2,1}) from the predicted contact map and calculate the precision, recall and F1 scores of these values, where:
根据标准的CASP定义,当两个Cβ原子之间的残基间距≤8.0 埃时,这些实验中的蛋白质残基被认为是接触的。对于长度为L的蛋白质,我们首先将其分为短(12>| i−j |≥6)、中等(24>| i−j |≥12)和长(| i−j |≥24)序列位置分离残基。从这些组中,我们从预测的接触图中取前L/k最高排名的预测(其中k∈{10,5,2,1}),计算这些值的精度、召回率和F1分数。
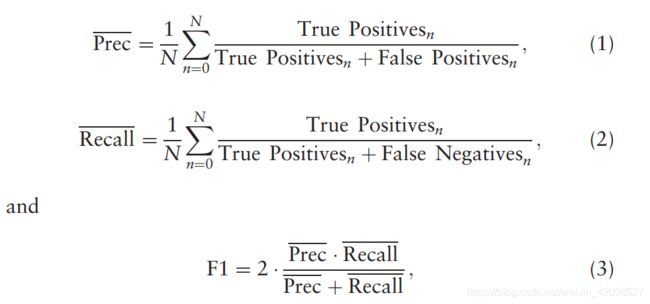
where N is the number of proteins in the test set.
其中N是测试集中的蛋白质数量。
These three metrics are chosen for consistency with the metrics used in the CASP12 rankings. However, these CASP metrics provide an evaluation on the positive predictions of a predictor, but do not provide any information on the predictions not in the top L/k predictions, especially those negative predictions. Thus, they are biased towards predictors which weight positive predictions. It should be noted that SPOT-Contact was not trained with a scaling factor for the positive samples. To analyze all the predictions from our model, we utilize the Area Under the receiver operating characteristic Curve (AUC) metric as an overall performance evaluator. This metric provides the probability that the predictor will rank a random positive sample higher than a random negative sample (Fawcett, 2006). We can compare the AUC scores of other predictors with respect to SPOT-Contact using a P-value, which indicates the statistical significance of the difference between the two predictors’ results (Hanley and McNeil, 1982). The smaller the P-value is, the more significant the difference between the two predictors. The AUC values for short-, medium- and long-range contacts were also obtained separately to examine the improvements in more detail.
为了一致性,我们选择了CASP12排名中的3个指标。然而,这些CASP指标提供了一个预测值的正预测值,但不提供任何不在前L/k预测值中的预测值的信息,尤其是那些负预测值。因此,他们偏向于那些对正样本预测加权的预测器。值得注意的是,对于阳性样本,SPOT-Contact没有使用比例因子进行训练。为了分析模型中的所有预测,我们利用AUC下的面积作为总体性能评估指标。这个指标提供了一个概率,即预测器将一个随机的正样本排序高于一个随机负样本。我们可以用P值来比较其他预测器的AUC得分与SPOT-Contact 的关系,这表明两个预测器的结果之间的差异具有统计学意义。P值越小,两个预测值之间的差异越显著。此外,还分别获得了短、中、长程接触的AUC值,以更详细地检查改进情况。
The latest CASP publication (Schaarschmidt et al., 2018) noted that the area under the Precision-Recall (PR) curve provides a balanced metric. Thus we also analyze the AUC of the PR curve, to analyze the positive predictions of the predictor.
最新的CASP出版物指出,精确召回(PR)曲线下的面积提供了一个平衡的指标。因此我们还分析了PR曲线的AUC,分析了预测器的正预测。
2.5 Method comparison
In order to gauge the performance of SPOT-Contact on our independent test sets, we chose several other readily-available, recently-developed contact-map predictors. The standalone versions of MetaPSICOV (Download: http://bioinfadmin.cs.ucl.ac.uk/downloads/MetaPSICOV/), Gremlin (Download: http://gremlin.bakerlab.org), SVMCon (Download: http://scratch.proteomics.ics.uci.edu), SVMSeq (Download: http://zhanglab.ccmb.med.umich.edu/SVMSEQ), DeepConPred and DeepRCon (obtained from Xiong et al., 2017) and PlmDCA (Download: https://github.com/pagnani/PlmDCA) were used in these experiments. The standalone version of MetaPSICOV also provided the results of EVFold, PSICOV and CCMPred. We also submitted jobs to the online servers of DNCON2 (server URL: http://sysbio.rnet.missouri.edu/dncon2/), RaptorX-Contact (Server URL: http://raptorx.uchicago.edu/ContactMap/), R2C (Server URL: http://www.csbio.sjtu.edu.cn/bioinf/R2C/), NeBcon (Server: https://zhanglab.ccmb.med.umich.edu) and CMapPro (Server URL: http://scratch.proteomics.ics.uci.edu/). Other predictors were considered, but were ultimately far too time-consuming to do large-scale predictions or were unavailable (Li et al., 2016).
为了在我们的独立测试集上测试SPOT-Contact的性能,我们选择了其他几个现成的,最近开发的接触图预测器。MetaPSICOV的独立版本(Download: http://bioinfadmin.cs.ucl.ac.uk/downloads/MetaPSICOV/), Gremlin (Download: http://gremlin.bakerlab.org), SVMCon (Download: http://scratch.proteomics.ics.uci.edu), SVMSeq (Download: http://zhanglab.ccmb.med.umich.edu/SVMSEQ), DeepConPred and DeepRCon (obtained from Xiong et al., 2017) and PlmDCA (Download: https://github.com/pagnani/PlmDCA) 被用于本次实验中。MetaPSICOV的独立版本还提供了EVFold、PSICOV和CCMPred的结果。我们还向DNCON2的在线服务器提交了JOB(server URL: http://sysbio.rnet.missouri.edu/dncon2/), RaptorX-Contact (Server URL: http://raptorx.uchicago.edu/ContactMap/), R2C (Server URL: http://www.csbio.sjtu.edu.cn/bioinf/R2C/), NeBcon (Server: https://zhanglab.ccmb.med.umich.edu) and CMapPro (Server URL: http://scratch.proteomics.ics.uci.edu/)。其他预测器也被考虑在内,但最终过于耗时,无法进行大规模预测或无法获得。
3 Results
The results of SPOT-Contact on each of the validation and independent testing datasets are shown in Supplementary Table S2 for AUC and the mean precision of the results when separated into both sequence separation cutoffs (short, medium and long), and top-ranking prediction cutoffs (L∕10, L∕5, L∕2 and L). The F1 results are provided in Supplementary Table S3. Here, we did not perform multi-fold cross-validation in training because of the time-consuming nature of every training run. Nevertheless, the comparable performance in AUC (0.976 versus 0.973) for the validation and the independent Test set indicate the robustness of the ensemble predictor. Similar precisions are observed regardless of whether it is a short, medium, or long-range contact (88%, 88% and 92% for top L∕10 predictions for short, medium and long-range contacts in the test set, respectively). The predictions of SPOT-Contact on the harder subset (Test-Hard) obtained somewhat lower AUC, precisions and F1 scores. This was expected, as more proteins in this subset have fewer homologous sequences and thus are harder to predict. The average number of effective homologous sequences from HHblits is 7.93 for the Test set but only 6.19 for the Test-Hard set.
3.1 Ensemble model analysis
To illustrate the contribution of each individual predictor, the individual and cumulative precision values for the Test-Hard are shown in Supplementary Tables S4 and S5, respectively. Because it is not possible to directly compare the ResNets used in RaptorX-Contact, we trained a pure ResNet model to compare to other ensemble component models. As the results in Supplementary Table S4 show, while the ResNet model is somewhat effective at short-range prediction, it lacks the long-range modeling capabilities to predict the long-range contacts as effectively as the pure 2D-BRLSTM model. However, ResNets and 2D-BRLSTMs can be enhanced by combining the two in hybrid models, showing the benefits of using the ResNets to congregate the short-term relationships and then using the 2D-BRLSTM to propagate this information throughout the protein image.
Supplementary Table S5 shows how much of a boost to performance each network adds to the original base network, culminating in an increase in the long-range predictions’ precisions by 5.31, 4.79, 5.24 and 4.54% for Test-Hard at the cutoffs of top L∕10, L∕5, L∕2, L prediction, respectively. While further gains could be obtained from adding more models, the cumulative performance gains become incrementally smaller as the number of networks in the ensemble increases, indicating that it would not be worthwhile to increase the number of networks further.
3.2 Feature importance
It is of interest to see the effect of the individual feature groups on the prediction accuracy. Much research has been conducted regarding the effectiveness of evolutionary profiles, sequence structure and physicochemical properties on protein structure prediction (Hanson et al., 2017), but such insight does not exist for the 2D features in protein contact map prediction. Thus, we have trained our baseline model without our 2D feature groups (the evolutionary coupling features and CCMpred output) sequentially to see the effect of the 2D features on our predictions. As is shown in Supplementary Table S6, 2D features from CCMpred and DCA led to significant improvement over the model based on 1D feature only. The improvement is significant in all short, medium and long-range contact pairs. For example, the improvement for top L/10 prediction is 3.7%, 6.5% and 10.3% for short, medium and long-range contacts, respectively. Similar level of improvement in F1 measure is also observed in Supplementary Table S7. The overall improvement in AUC is 3.3% from 0.908 to 0.941.
3.3 Comparison to the 17 other methods
The performance of the 17 other predictors on a subset of Test-Hard can be found in Supplementary Table S8 through Supplementary Table S11. The corresponding Receiver-Operating Characteristic (ROC) and PR curves are shown in Figures 2 and 3. Mean precisions given by different methods for short, medium and long-range contacts are also shown in Figure 4. Predictors such as CMapPro and PSICOV have maximum length or a minimum number of sequence homologues requirements, which trimmed the size of our dataset from 277 to 228. SPOT-contact significantly outperforms all compared models over all performance evaluations on this dataset. For example, as shown in Supplementary Table S10, the largest AUC is 0.958 given by SPOT-contact, followed by SPOT-Contact-2016 (0.950), RaptorX-Contact (0.909) and DNCON2 (0.886). The AUC values for all other methods are less than 0.84. A two-tailed P-value of < 10–6 is obtained when comparing SPOT-contact’s AUC value against all external predictors. This improvement is not dependent on the length cutoff, as Supplementary Table S10 shows that the increase of AUC for SPOT-Contact is present over all residue-separation cutoffs. For example, the AUC values for short-, medium- and long-range contacts are 0.935, 0.949 and 0.888 by RaptorX-Contact, respectively, compared to 0.960, 0.965 and 0.951 by SPOT-Contact, respectively.
We also compare the PR curves of each predictor, and calculate the AUC of these curves in Supplementary Table S11. SPOT-Contact obtains the highest AUC, even when segmented by residue separation. SPOT-Contact increases on RaptorX-Contact’s overall PR AUC by 0.1 from 0.554 to 0.658, 0.076 from 0.584 to 0.660 and 0.07 from 0.457 to 0.527 for the short-, medium- and long-range contacts, respectively.
Most significantly, SPOT-contact increases the already-outstanding long-range contact precision scores of RaptorX-contact (e.g. from 51.1 to 55.6 in top L long-range prediction), further increasing the gap between modern machine learning techniques and ECA methods. This is independent of sequence profile database selection, as SPOT-Contact-2016 also improved on RaptorX-Contact in each analyzed metric.
In particular, SPOT-contact is the only method to achieve more than 80% precision for short, medium and long-distance contacts for top L/10 predictions. This happened without specific training for precision. It can be noticed that the ECA-based predictors perform poorly on this dataset, due many proteins in this dataset not having a large number of sequence neighbors in the existing sequence library. SPOT-contact receives slightly higher evaluation metrics on this subset of 228 proteins in Test-Hard than the results in Supplementary Table S2 because the maximum length and minimum number of sequence homologue restrictions placed by other predictors makes this subset easier to predict than the full 277-protein Test-Hard dataset. This is confirmed by a similar decrease in the evaluation metrics from RaptorX-Contact; for instance the mean precision of the top L long-range residue pairs decreases from 51.09% in the subset to 49.31% in the full set.
To confirm the dependence of method-performance on the number of homologous sequences, we present the mean precision values of the top L/5-ranked predictions as a function of the maximum cumulative Neff values (the number of effective homologous sequences, ranging from 1 to 20) from HHblits, in Supplementary Figure S1. All methods had their lowest performance for lower Neff sequences. SPOT-Contact improves over RaptorX-Contact at all Neff values, with the largest improvement for low-medium range Neff sequences. We further analyze the results in accordance with the number of contacts a residue has from the reduced Test-Hard. We bin each residue in our database depending on the number of contacts it has, and calculate the mean precision of the top L/5 long-range precisions. As shown in Supplementary Figure S2, residues with fewer contacts (surface contacts) are much harder to discriminate from their non-contacts, with each additional contact bringing an almost linear increase in performance to all predictors. SPOT-Contact and RaptorX-Contact show a distinct advantage over all other methods, with our method maintaining an increase in performance over RaptorX-Contact for all contact numbers.
We further examined the dependence of prediction precision on protein secondary structure. Supplementary Table S12 compares the performance of the four top-performing methods (metaPSICOV, RaptorX-Contact, SPOT-Contact-2016 and SPOT-Contact) for residues with different secondary structure elements on the full set of Test-Hard. The contacts between sheet residues have the highest precision for all three methods. Using the updated sequence profiles, SPOT-Contact increases on SPOT-Contact-2016 over all secondary structure pairs for all length cutoffs. SPOT-Contact also increases upon the performance of RaptorX-Contact, which attained similar performance for some residue pairs with SPOT-Contact-2016.
3.4 CASP results
For completeness, we compared SPOT-Contact the other predictors in Supplementary Table S13 for the available TFM CASP targets (22 proteins only). At the time of testing, several servers were not available and others are unable to predict the full 22-protein set. Only the remaining servers were provided in Supplementary Table S13. SPOT-Contact achieves the highest AUC of both the ROC and PR curves (0.906 compared to the next highest 0.862, and 0.443 compared to 0.369, respectively), and also achieves the highest mean precision values across all length cutoffs and distance separations. For example, the L/5 cutoff scores for RaptorX-Contact (the CASP12 winner) and SPOT-Contact are 59.76% and 64.06%, 56.14% and 64.41% and 58.99% and 63.95% for the short-, medium- and long-range contacts, respectively.
4 Conclusion
In this paper, we have developed a new predictor called SPOT-Contact for protein contact maps. This method is built on the previous success of residual CNN in contact map prediction by RaptorX-Contact and the capability of capturing long-range interactions by LSTM-RNN networks by inputting whole sequences. In addition, the ensemble of six predictors with different combinations of networks removes prediction noise and makes prediction more generalizable. Using 228 recently-solved structures as an independent test set, SPOT-Contact is consistently more accurate in contact prediction across contacts at different sequence separations (Fig. 4), across proteins with different number of effective homologous sequences (Supplementary Fig. S1) and across residues with different number of contacts (Supplementary Fig. S2). The improvement in AUC is 5% over the next best technique RaptorX-Contact, and is statistically significant (P < 10–6). The result highlights the usefulness of coupling ResNets with two-dimensional LSTM networks.
在这篇论文中,我们开发了一种新的预测器,称为蛋白质接触图的SPOT-Contact。该方法是基于残差CNN在 RaptorX-Contact的接触图预测中的成功,以及LSTM-RNN网络通过输入全序列捕捉远距离相互作用的能力。另外,六个不同网络组合的预测器的组合消除了预测噪声,使预测更具普遍性。使用228个最近解决的结构作为一个独立的测试集, SPOT-Contact在不同序列间隔的接触预测中始终更准确(图4),具有不同数量的有效同源序列的跨蛋白质(补充图S1)和具有不同接触数的跨残基(补充图S2)。与次优技术RaptorX-Contact相比,AUC的改善为5%,具有统计学意义(P < 10–6)。结果表明了ResNets与二维LSTM网络耦合的有效性。
————————————————————————————
Result部分未翻译!!!!!