R -- 层次聚类和划分聚类
brief
聚类分析是一种数据归约技术,旨在揭漏一个数据集中观测值的子类。子类内部之间相似度最高,子类之间差异性最大。至于这个相似度是一个个性化的定义了,所以有很多聚类方法。
最常用的聚类方法包括层次聚类和划分聚类。
- 层次聚类,每一个观测自成一个类,然后这些类两两合并,直到所有的类都被合并为止。计算相似度的方法有单联动,全联动,平均联动,质心和ward法。
- 划分聚类,首先指定子类个数K,然后观测被随机分配成K类,再重新聚合成为类。计算相似度的方法包括K-means和围绕中心点的划分PAM
每个聚类方法都有优缺点,甚至聚类的时候出现不存在的类,这些方法最好是对照使用。
基本步骤
有效的聚类分析是一个多步骤的过程,每一步的决策都会影响下一步的分析。所以多探索多尝试最重要。
基本步骤:
- 选择合适的变量。这些变量对数据的理解和识别区分应该起主要作用,否则再高级的聚类方法也无法弥补聚类变量选不好的问题。
- 缩放数据。如果数据的波动范围很大,那么该变量对结果的影响也是最大的(这不就是合适的变量嘛)。但用这样的原始数据并不可取,最好用标准化的数据代替。
下面是三种缩放方法:
df1 <- apply(mydata, 2, function(x){(x-mean(x))/sd(x)})
df2 <- apply(mydata, 2, function(x){x/max(x)})
df3 <- apply(mydata, 2, function(x){(x ̢ mean(x))/mad(x)})
- 寻找异常点。很多聚类方法对异常值很敏感,他能扭曲我们的聚类方案。可以通过outliers包中的函数检测离群点,mvoutlier包中有识别多元变量的离群点函数。
- 计算距离。dist()计算距离和选择相应的算法
- 选择聚类算法。小样本(<150个观测)或者嵌套聚类可以选择层次聚类。划分聚类可以处理大样本但是需要先确定子类个数K。
- 尝试其他的聚类算法或者改变距离计算方法
- 确定类的数目。可以使用NbCluster包中的NbCluster()函数,该函数提供了30个不同的指标帮助你进行选择。
- 提取子类
- 结果可视化
- 解读子类。子类内部有何相似性?子类之间有何差异性?
- 结果验证。有没有可能是数据集或者聚类方法导致了这种结果,事实上这种划分是没有意义的?
其中距离的计算部分:

其他计算距离的算法详见帮助文档
?dist
层次聚类



# install.packages("flexclust")
data(nutrient, package="flexclust")
row.names(nutrient) <- tolower(row.names(nutrient))
head(nutrient)
# 缩放数据
nutrient.scaled <- scale(nutrient,center = T,scale = T)
head(nutrient.scaled)
# 计算欧几里得距离
d <- dist(nutrient.scaled)
head(d)
typeof(d)
str(d)
# 将距离数据传入层次聚类函数
# 距离数据
fit.average <- hclust(d, method="average")
plot(fit.average, hang=-1, cex=.8, main="Average Linkage Clustering")


- 上述的层次聚类树可视化了聚类的过程,观测之间的相似性和异质性也可以清楚的看到。接下来我们还可以进行子类的确定,让观测分配到合适的类群。最后进行子类提取
library(NbClust)
# 需要输入需要聚类的数据框,距离计算公式,聚类方法
nc <- NbClust(nutrient.scaled, distance="euclidean",
min.nc=2, max.nc=15, method="average")
table(nc$Best.n[1,]) # 看看多少个指标支持对应的子类个数
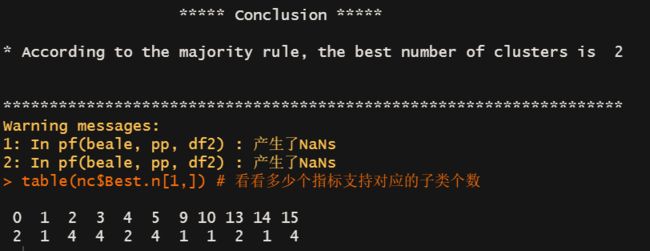
有1个指标支持1个子类
有4个指标支持2个子类
有4个指标支持3个子类
有4个指标支持5个子类
Nbcluster推荐聚成2类
# 那就把K设置成 2或者3或者5试试看
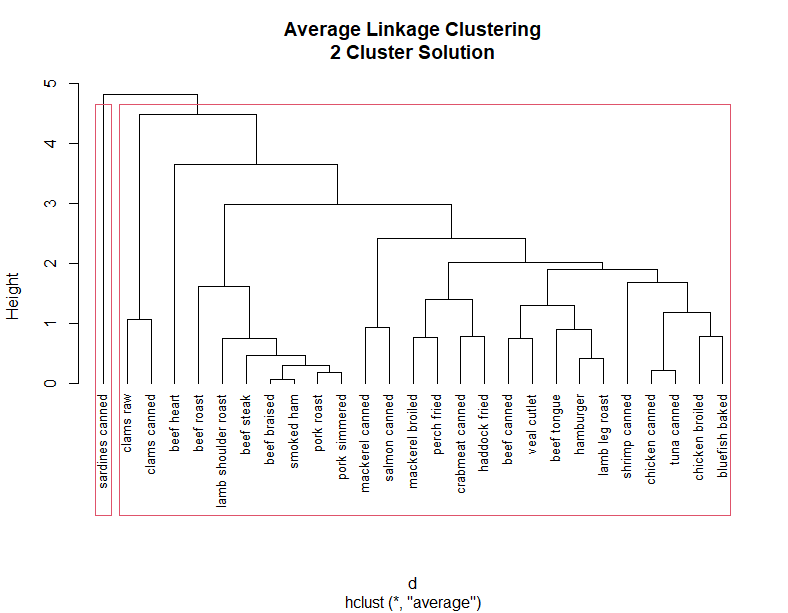
clusters <- cutree(fit.average, k=2) # cutree将层次聚类结果划分为5个部分
# 第一个子类包括了26个观测,第二个子类包括了一个观测
table(clusters)
# 利用聚合函数看看原来的数据框
aggregate(nutrient, by=list(cluster=clusters), median) # 主要是calcium的含量区别
# 可视化看看
plot(fit.average, hang=-1, cex=.8,
main="Average Linkage Clustering\n2 Cluster Solution")
rect.hclust(fit.average, k=2)
划分聚类
kmeans
brief

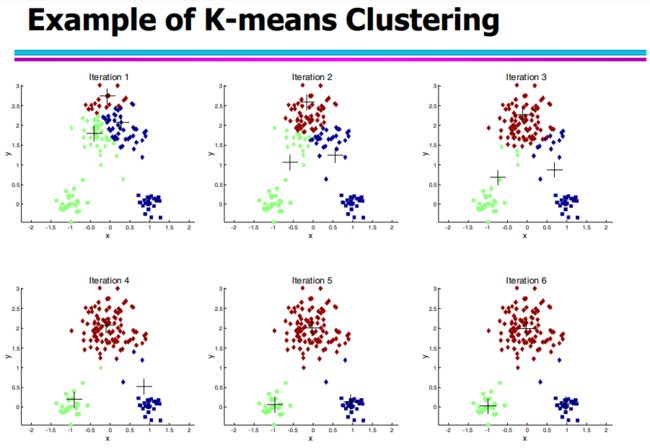


实例
- 第一步:确定子类个数K值
library(NbClust)
data(wine, package="rattle")
head(wine)
df <- scale(wine[-1])
set.seed(1234)
nc <- NbClust(df, min.nc=2, max.nc=15, method="kmeans")
table(nc$Best.n[1,]) # 3个子类最好
# 或者使用碎石图的方法找拐点,如下代可以实现
wssplot <- function(data, nc=15, seed=1234){
wss <- (nrow(data)-1)*sum(apply(data,2,var))
for (i in 2:nc){
set.seed(seed)
wss[i] <- sum(kmeans(data, centers=i)$withinss)}
plot(1:nc, wss, type="b", xlab="Number of Clusters",
ylab="Within groups sum of squares")
}
wssplot(df)
- 第二步:聚类
set.seed(1234)
fit.km <- kmeans(df, 3, nstart=25)
str(fit.km)

size : 每个子类包含的观测数目
iter : 迭代次数
withinss : 子类内部 距离之和
centers: 子类的中心点,都少个变量就会有对应的多少个数值,组成了一个高维的点。可能不好理解,看看看下面的代码。
fit.km$centers
aggregate(wine[-1], by=list(cluster=fit.km$cluster), mean)

第一个子类的中心点是![]()
- 结果怎么样?
wine$ type是真实的分类,fit.km$ cluster是kmeans的聚类
可以看到大约6个观测被错误的分配了,三个观测属于第二个子类,却被分到了第一个子类,还有三个观测属于第二个子类,却被分到了第三个子类。

PAM
brief


实例
library(cluster)
set.seed(1234)
fit.pam <- pam(wine[-1], k=3, stand=TRUE)
fit.pam$medoids
clusplot(fit.pam, main="Bivariate Cluster Plot")
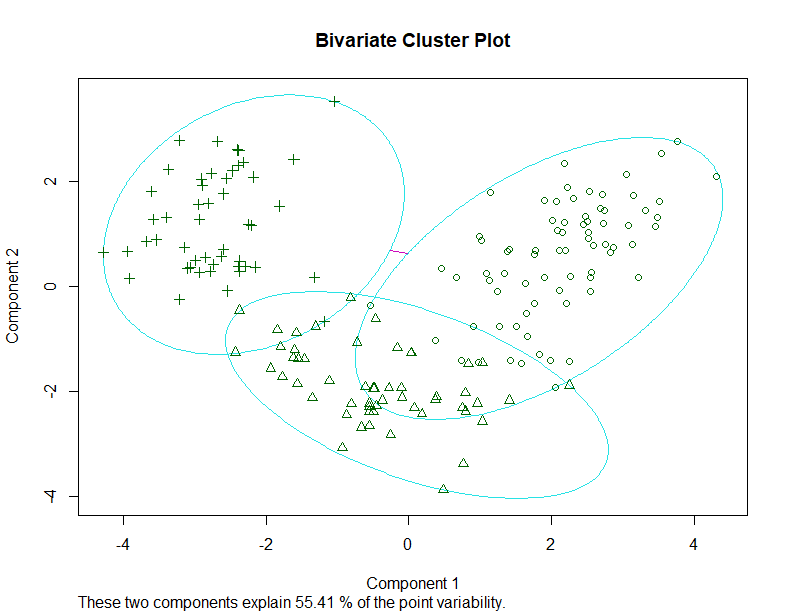
- 结果怎么样
table(wine$Type,fit.pam$clustering)

在这个数据集上PAM表现不如Kmeans。所以数据集和算法相互影响的,每种聚类方法都有优缺点,多对照使用。