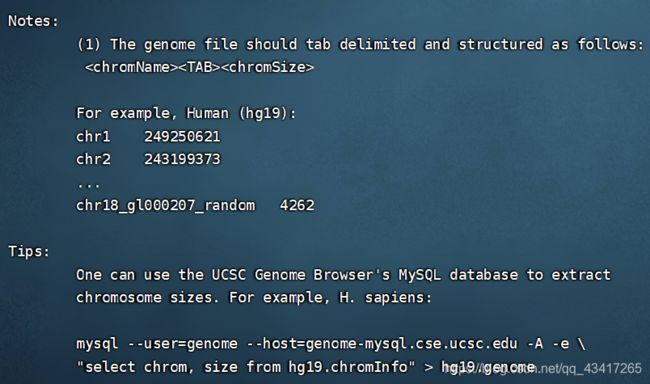
- 推荐一份生物信息学入门很好的参考材料
小明的数据分析笔记本
链接是https://bioinformatics.uconn.edu/resources-and-events/tutorials-2/这个是康涅狄格大学(UniversityofConnecticut)提供的一份教程,主要的内容包括1、生物信息学中经常用到的文件格式image.png2、linux操作系统和R语言的基础知识image.png3、转录组数据的处理流程image.png这里包括有参
- 【机器学习】朴素贝叶斯方法的概率图表示以及贝叶斯统计中的共轭先验方法
Lossya
机器学习概率论人工智能朴素贝叶斯共轭先验
引言朴素贝叶斯方法是一种基于贝叶斯定理的简单概率模型,它假设特征之间相互独立。文章目录引言一、朴素贝叶斯方法的概率图表示1.1节点表示1.2边表示1.3无其他连接1.4总结二、朴素贝叶斯的应用场景2.1文本分类2.2推荐系统2.3医疗诊断2.4欺诈检测2.5情感分析2.6邮件过滤2.7信息检索2.8生物信息学三、朴素贝叶斯的优点四、朴素贝叶斯的局限性4.1特征独立性假设4.2敏感于输入数据的表示4
- 零基础入门生信数据分析——导读
呆猪儿
生信之转录组——上游分析生信之转录组——下游分析学习方法r语言数据分析数据库数据挖掘需求分析大数据
零基础入门生信数据分析——导读生信数据分析,即生物信息学数据分析,是一个涵盖了生物学、计算机科学、数学和统计学等多个领域的交叉学科。它主要利用计算机算法和统计方法对生物学数据进行处理、分析和解释,以揭示生物分子、细胞、组织和生物体等各个层次的生物学规律和机制。本帖主要是为生信数据分析的各个分析点提供跳转链接(简单说就是提供了一个目录供大家选择自己想要的知识点可以直接跳转)关联的生信数据分析的分析点
- NCBI BLAST+:分析生物内在编码的工具
belldeep
生物信息学Blast生物数据分析
在生物信息学的广阔领域中,NCBI(NationalCenterforBiotechnologyInformation,美国国立生物技术信息中心)开发的BLAST(BasicLocalAlignmentSearchTool,基本局部比对搜索工具)无疑是一把不可或缺的分析工具。NCBIBLAST+,作为其最新版本2.16.0+,为科研工作者提供了一套强大的序列比对和搜索功能,帮助解析生命现象背后的遗
- 【图论简介】
WA-自动机
图论深度优先算法架构后端前端面试
图论简介图论是一门数学分支,主要研究图(Graph)的性质、结构和应用。图论在计算机科学、网络理论、优化问题、生物信息学等多个领域都有广泛的应用。本文将简要介绍图论的基本概念、常见算法及其在实际中的应用。一、图的基本概念图(Graph):图是由一组顶点(Vertices)和连接顶点的边(Edges)组成的结构。可以表示为(G=(V,E)),其中(V)是顶点的集合,(E)是边的集合。根据边的不同属性
- 生信圆桌:专业生信服务器与平台服务的提供者
生信圆桌x生信云服务器
服务器人工智能运维
生信圆桌是一个专注于提供生物信息学(生信)服务器和平台服务的领先企业,致力于为全球科研机构、企业和独立研究者提供高性能的生信分析解决方案。随着生物信息学研究对计算资源的需求日益增加,生信圆桌凭借其先进的服务器技术和专业的服务团队,成为了生信领域中不可或缺的合作伙伴。访问生信圆桌,使用生信云。高效分析少走弯路www.tebteb.cc生信圆桌的核心服务高性能生信服务器定制:生信圆桌为客户提供定制化的
- 用Python实现生信分析——基序(Motif)识别详解
写代码的M教授
生信分析python开发语言
1.什么是基序(Motif)?在生物信息学中,基序(Motif)是指在生物序列(如DNA、RNA或蛋白质序列)中具有特定功能或结构的短序列片段。基序通常在生物进化中得到保留,因为它们在生物学功能中起着重要作用。例如,在DNA序列中,基序可能是一个转录因子结合位点;在蛋白质序列中,基序可能是一个具有特定功能的结构域。基序识别是指从一组生物序列中识别出保守的短序列片段,这对于功能预测、基因调控网络分析
- 数据结构与算法——动态规划
passion更好
数据结构C++动态规划算法
目录引言最优子结构重叠子问题打家劫舍(LeetCode198题)经典例题1.爬楼梯(LeetCode70题)2.斐波那契数列(LeetCode126题)3.最长公共子序列(LeetCode95题)引言动态规划(DynamicProgramming,简称DP)是一种在数学、计算机科学、经济学和生物信息学等领域广泛使用的算法设计技术。它通过把原问题分解为相对简单的子问题的方式,来求解复杂问题。动态规划
- 深度学习——概念引入
韶光流年都束之高阁
深度学习日记深度学习人工智能职场和发展
深度学习深度学习简介深度学习分类根据网络结构划分:循环神经网络卷积神经网络根据学习方式划分:监督学习无监督学习半监督学习根据应用领域划分:计算机视觉自然语言处理语音识别生物信息学深度学习简介深度学习(DeepLearning,DL)是机器学习领域中的一个新的研究方向,主要是通过学习样本数据的内在规律和表示层次,让机器能够具有类似于人类的分析学习能力。深度学习的最终目标是让机器能够识别和解释各种数据
- 考研调剂:中医生命科学
菌心说双脑论
科学网—考研调剂——欢迎研究生调剂到我们的招生专业方向“中西医结合基础”:中医药与肠道菌群、生物信息学等交叉学科-张成岗的博文http://blog.sciencenet.cn/home.php?mod=space&uid=40692&do=blog&id=1281078欢迎各位有志于从事中医生命科学、解码中医、中西医结合以及医学与数学、计算机科学等交叉学科研究的青年才俊加入我们的研究团队,共同见
- 2020-04-07
liuyang2020
学习小组Day2笔记--linux入门(刘阳)1.为什么学习linux大多数人用的是可视化界面,便捷的windows,linux用户量比较少,但是需要知道,linux的功能相当的强大,对于数据处理、程序运行方面的优势,那是其它的系统无法比拟的,生物信息学数据处理对电脑要求较高,因此学习linux,,嘿嘿,大势所趋。2.linux操作2.1登录远程登录linux服务器,好像有很多连接软件,今天尝试应
- Python在生物信息学中的应用:有序字典
简说基因-专业生信合作伙伴
python开发语言
我们知道,通过{}创建的字典是无序的。如何创建有序字典呢?解决方案可以使用collections模块中的OrderedDict类。当对字典做迭代时,它会严格按照元素添加的顺序进行。例如:from collection import OrderedDictd=OrderedDict()d['1st'] = 1d['2nd'] = 2d['3rd'] = 3d['4th']=4forkeyind:
- Python在生物信息学中的应用:同时对数据做转换和换算
简说基因-专业生信合作伙伴
python开发语言
我们需要调用一个换算(reduction)函数,例如sum()、min()、max()等,但首先得对数据做转换或筛选。解决方案一种优雅的方式能将数据换算和转换结合在一起,即在函数中使用生成器表达式。例如,要计算平方和,可以这样:nums=[1,2,3,4,5]s=sum(x*xforxinnums)更多的例子:#Determineifany.pyfilesexistinadirectoryimpo
- Python在生物信息学中的应用:列表推导式
简说基因-专业生信合作伙伴
pythonwindows开发语言
列表中有一些数据,我们想提取或删除某些值,该怎么办?解决方案最简单的方法是使用列表推导式(listcomprehension)。例如:>>>mylist=[1,4,-5,10,-7,2,3,-1]>>>[nforninmylistifn>0][1,4,10,2,3]>>>[nforninmylistifn>>列表推导式的使用需要注意其内存占用,当原始列表比较大时,其内存占用较高,可以使用生成器表达
- 最长公共子序列(LCS)
算法
定义(维基百科)在一个序列集合中(通常为两个序列)查找所有序列中最长的子序列。这与查找最长公共子串的问题不同的地方是:子序列不需要在原序列中占用连续的位置。最长公共子序列问题是一个经典的计算机科学问题,也是数据比较程序,比如Diff工具和生物信息学应用的基础。它也被广泛地应用在版本控制,比如Git用来调和文件之间的改变解决方案这类问题通常都是采用动态规划的思想来解决,核心就是构造出动态解决方程。以
- 自学生物信息学
gtt儿_生物信息学习
我是生物工程专业出身,在大三保研时选择了生物信息的道路,到现在为止已经在行业里摸爬滚打了6年的时间,在这6年的学习之路上疑惑过,也迷茫过,特此把我学习的过程以及遇到的问题总结出来以让大家避免出现同样的问题。在我学习生物信息过程的基础上带着大家顺畅的走一遍。在学习生物信息学之前,我们先来了解一下什么是生物信息学。生物信息学,顾名思义,生物学和信息学的结合。生物学,这个对大家比较简单,基本入生信行的同
- 我们能成为孩子的上帝吗—— 谁来管理非法行医的贺建奎
闲月农
贺建奎,原南方科技大学副教授,毕业于美国斯坦福大学,拥有多学科交叉的背景,并在基因测序仪研究,CRISPR基因编辑,生物信息学等多个领域取得研究突破。2018年11月26日,贺建奎“基因编辑婴儿”事件引发轩然大波。2018年12月19日,贺建奎入选《Nature》年度十大科学人物。2019年4月18日,上榜美国《时代》杂志(Time)2019年度全球百位最具影响力人物榜单。2019年12月30日,
- 2022-01-27
学习生信的小兔子
参考:生物信息学100个基础问题——第1~5题答案公布-知乎(zhihu.com)掌握FASTQ格式特点第2行就是测序得到的序列信息,一般用ATCGN来表示,其中N用于荧光信号干扰无法判断到底是哪个碱基时的代表符号;第3行以“+”开始,可以储存一些附加信息,但目前的测序fastq文件这一行一般是空的。第4行储存的是质量信息,与第2行的碱基序列是一一对应的,其中的每一个符号对应的ASCII值是经过换
- 金域医学:医检行业顶级学术委员会成立,钟南山院士任主席
里昂杰森
4位院士领衔23位顶级专家加盟,金域医学“最强大脑”助力中国医学检验2017年12月1日,国内第三方医学检验行业的开拓者和引领者广州金域医学检验集团在广州国际生物岛总部,召开金域医学学术委员会成立大会暨金域学术汇报会由呼吸系统疾病专家、中国工程院院士钟南山出任委员会主席,医学遗传学家、中国工程院院士曾溢滔,生物信息学家、中国科学院院士陈润生,以及我国著名肾脏病专家、中国科学院院士侯凡凡出任委员会顾
- 机器学习系列——(十九)层次聚类
飞影铠甲
机器学习机器学习聚类人工智能
引言在机器学习和数据挖掘领域,聚类算法是一种重要的无监督学习方法,它试图将数据集中的样本分组,使得同一组内的样本相似度高,不同组间的样本相似度低。层次聚类(HierarchicalClustering)是聚类算法中的一种,以其独特的层次分解方式,在各种应用场景中得到广泛应用,如生物信息学、图像分析、社交网络分析等。一、概述层次聚类算法主要分为两大类:凝聚的层次聚类(AgglomerativeHie
- 东南大学-生物信息学
wangchuang2017
http://www.lmbe.seu.edu.cn/chenyuan/xsun/bioinfomatics/Web/Index.html目录image第1章生物信息学引论第2章生物信息学的生物学基础第3章序列比较第4章生物分子数据库第5章基因组信息分析第6章系统发生分析第7章蛋白质结构预测第8章基因表达数据分析附录常用基本词汇表
- TCGA新版数据库表达矩阵提取
医学和生信笔记
本文首发于公众号:医学和生信笔记医学和生信笔记,专注R语言在临床医学中的使用,R语言数据分析和可视化。主要分享R语言做医学统计学、meta分析、网络药理学、临床预测模型、机器学习、生物信息学等。现在使用TCGAbiolinks下载转录组数据后,直接是一个SummarizedExperiment对象,这个对象非常重要且好用。因为里面直接包含了表达矩阵、样本信息、基因信息,可以非常方便的通过内置函数直
- R语言可视化学习笔记之ggridges包
生信宝典
R生物信息生物信息可视化
作者:严涛浙江大学作物遗传育种在读研究生(生物信息学方向)伪码农,R语言爱好者,爱开源。严涛老师的绘图教程还有:gganimate|诺奖文章里面的动图布局教程来了!!ggplot2学习笔记之图形排列R包ggseqlogo|置换序列分析图ggplot2高效实用指南(可视化脚本,工具,套路,配色)简介ggridges。主要包用来绘制山峦图产品尤其的英文针对时间或者空间分布****可视化。具有十分好的效
- microRNA数据库与预测、功能分析软件大全
Seurat_Satija
在microRNA的研究中,生物信息学发挥越来越重要的作用,以下是microRNA相关的数据库与预测、功能分析软件,绝对值得收藏。1.miRBase:http://www.mirbase.orgmiRBase序列数据库是一个提供包括已发表的miRNA序列数据、注释、预测基因靶标等信息的全方位数据库,是存储miRNA信息最主要的公共数据库之一。miRBase提供便捷的网上查询服务,允许用户使用关键词
- 从列表中删除元素|自学生信Python(第十六天)
天明豆豆
从列表中删除元素Python有从数据结构对象,如列表和字典中去除数据项的函数。写在前面的话:本人是一枚生物学的学生,由于对生物信息学特别感兴趣,于是想自学生物信息学(新手莫怪)。了解到生物信息学要有编程基础,尤其是要会一门编程语言,例如:R语言、Python、Perl等,还要熟悉Linux系统,作为生信小白,听说Python挺简单的,于是就自学了Python,花了两天时间了解了Python的基础语
- 「转录组」从环境配置之conda
旮旯蜗牛_c299
image.png什么是condaconda:开源包管理系统和环境管理系统,用于安装多个版本的软件包及其依赖关系,并在它们之间轻松切换。系统:适用Linux,OSX和Windows。For:为Python程序创建的,但可以打包和分发任何软件。【生物信息学频道bioconda】Anaconda是一个开源的Python发行版本,包含了conda、python等180多个科学包及其依赖项。因为包含了大量
- 生信绘图:在线绘制 序列 Logo 图
Ningbo_JiaYT
统计绘图生物信息学R学习方法
本文介绍通过WebLogo网站在线绘制序列Logo图(序列分析图)。网站链接:WebLogo3-About(threeplusone.com)1序列Logo图序列Logo是一种常用于可视化DNA、RNA或氨基酸序列中保守性和模式的图形化方法。它是由生物信息学领域中的生物学家TomSchneider和R.MichaelStephens在1990年首次引入的。序列Logo通过显示序列中每个位置上不同碱
- 理解生物信息学FASTA格式
陈佶1
在生物信息学中,FASTA格式是一种基于文本的、用于表示核苷酸序列或氨基酸序列的格式。在这种格式中碱基对或氨基酸用单个字母来表示,且允许在序列前添加序列名及注释。FASTA文件以序列表示和序列作为一个基本单元,各行记录信息如下:第一行是由大于号">"开头的任意文字说明,用于序列标记,为了保证后续分析软件能够区分每条序列,单个序列的标识必须具有唯一性。;从第二行开始为序列本身,只允许使用既定的核苷酸
- Cytoscape软件下载、安装、插件学习[基础教程]
小杜的生信筆記
R语言精美图形绘制教程数据分析Cytoscape网络图富集分析信息可视化生物信息学r语言
写在前面今天分享的内容是自己遇到问题后,咨询社群里面的同学,帮忙解决的总结。关于Cytoscape,对于做组学或生物信息学的同学基本是陌生的,可能有的同学用这个软件作图是非常溜的,做出来的网络图也是十分的好看,“可玩性”很高,就像前面分享的aPEAR包一样aPEAR包绘制功能富集网络图。自己在前面写论文的时候也是一直在使用,以前使用的版本是3.3.0的版本。但是,时间一长,很多操作都忘记。今天,在
- 支持向量机
小森( ﹡ˆoˆ﹡ )
机器学习算法支持向量机算法机器学习
支持向量机(SupportVectorMachine,SVM)是一个非常优雅的算法,具有非常完善的数学理论,常用于数据分类,也可以用于数据的回归预测中。支持向量机在许多领域都有广泛的应用,如文本分类、图像识别、生物信息学、金融预测等。支持向量机的应用:(1)文本分类:支持向量机可以用于文本分类任务,如垃圾邮件过滤、情感分析、主题分类等。通过对文本数据进行预处理,提取特征,然后使用支持向量机进行训练
- java观察者模式
3213213333332132
java设计模式游戏观察者模式
观察者模式——顾名思义,就是一个对象观察另一个对象,当被观察的对象发生变化时,观察者也会跟着变化。
在日常中,我们配java环境变量时,设置一个JAVAHOME变量,这就是被观察者,使用了JAVAHOME变量的对象都是观察者,一旦JAVAHOME的路径改动,其他的也会跟着改动。
这样的例子很多,我想用小时候玩的老鹰捉小鸡游戏来简单的描绘观察者模式。
老鹰会变成观察者,母鸡和小鸡是
- TFS RESTful API 模拟上传测试
ronin47
TFS RESTful API 模拟上传测试。
细节参看这里:https://github.com/alibaba/nginx-tfs/blob/master/TFS_RESTful_API.markdown
模拟POST上传一个图片:
curl --data-binary @/opt/tfs.png http
- PHP常用设计模式单例, 工厂, 观察者, 责任链, 装饰, 策略,适配,桥接模式
dcj3sjt126com
设计模式PHP
// 多态, 在JAVA中是这样用的, 其实在PHP当中可以自然消除, 因为参数是动态的, 你传什么过来都可以, 不限制类型, 直接调用类的方法
abstract class Tiger {
public abstract function climb();
}
class XTiger extends Tiger {
public function climb()
- hibernate
171815164
Hibernate
main,save
Configuration conf =new Configuration().configure();
SessionFactory sf=conf.buildSessionFactory();
Session sess=sf.openSession();
Transaction tx=sess.beginTransaction();
News a=new
- Ant实例分析
g21121
ant
下面是一个Ant构建文件的实例,通过这个实例我们可以很清楚的理顺构建一个项目的顺序及依赖关系,从而编写出更加合理的构建文件。
下面是build.xml的代码:
<?xml version="1
- [简单]工作记录_接口返回405原因
53873039oycg
工作
最近调接口时候一直报错,错误信息是:
responseCode:405
responseMsg:Method Not Allowed
接口请求方式Post.
- 关于java.lang.ClassNotFoundException 和 java.lang.NoClassDefFoundError 的区别
程序员是怎么炼成的
真正完成类的加载工作是通过调用 defineClass来实现的;
而启动类的加载过程是通过调用 loadClass来实现的;
就是类加载器分为加载和定义
protected Class<?> findClass(String name) throws ClassNotFoundExcept
- JDBC学习笔记-JDBC详细的操作流程
aijuans
jdbc
所有的JDBC应用程序都具有下面的基本流程: 1、加载数据库驱动并建立到数据库的连接。 2、执行SQL语句。 3、处理结果。 4、从数据库断开连接释放资源。
下面我们就来仔细看一看每一个步骤:
其实按照上面所说每个阶段都可得单独拿出来写成一个独立的类方法文件。共别的应用来调用。
1、加载数据库驱动并建立到数据库的连接:
Html代码
St
- rome创建rss
antonyup_2006
tomcatcmsxmlstrutsOpera
引用
1.RSS标准
RSS标准比较混乱,主要有以下3个系列
RSS 0.9x / 2.0 : RSS技术诞生于1999年的网景公司(Netscape),其发布了一个0.9版本的规范。2001年,RSS技术标准的发展工作被Userland Software公司的戴夫 温那(Dave Winer)所接手。陆续发布了0.9x的系列版本。当W3C小组发布RSS 1.0后,Dave W
- html表格和表单基础
百合不是茶
html表格表单meta锚点
第一次用html来写东西,感觉压力山大,每次看见别人发的都是比较牛逼的 再看看自己什么都还不会,
html是一种标记语言,其实很简单都是固定的格式
_----------------------------------------表格和表单
表格是html的重要组成部分,表格用在body里面的
主要用法如下;
<table>
&
- ibatis如何传入完整的sql语句
bijian1013
javasqlibatis
ibatis如何传入完整的sql语句?进一步说,String str ="select * from test_table",我想把str传入ibatis中执行,是传递整条sql语句。
解决办法:
<
- 精通Oracle10编程SQL(14)开发动态SQL
bijian1013
oracle数据库plsql
/*
*开发动态SQL
*/
--使用EXECUTE IMMEDIATE处理DDL操作
CREATE OR REPLACE PROCEDURE drop_table(table_name varchar2)
is
sql_statement varchar2(100);
begin
sql_statement:='DROP TABLE '||table_name;
- 【Linux命令】Linux工作中常用命令
bit1129
linux命令
不断的总结工作中常用的Linux命令
1.查看端口被哪个进程占用
通过这个命令可以得到占用8085端口的进程号,然后通过ps -ef|grep 进程号得到进程的详细信息
netstat -anp | grep 8085
察看进程ID对应的进程占用的端口号
netstat -anp | grep 进程ID
&
- 优秀网站和文档收集
白糖_
网站
集成 Flex, Spring, Hibernate 构建应用程序
性能测试工具-JMeter
Hmtl5-IOCN网站
Oracle精简版教程网站
鸟哥的linux私房菜
Jetty中文文档
50个jquery必备代码片段
swfobject.js检测flash版本号工具
- angular.extend
boyitech
AngularJSangular.extendAngularJS API
angular.extend 复制src对象中的属性去dst对象中. 支持多个src对象. 如果你不想改变一个对象,你可以把dst设为空对象{}: var object = angular.extend({}, object1, object2). 注意: angular.extend不支持递归复制. 使用方法: angular.extend(dst, src); 参数:
- java-谷歌面试题-设计方便提取中数的数据结构
bylijinnan
java
网上找了一下这道题的解答,但都是提供思路,没有提供具体实现。其中使用大小堆这个思路看似简单,但实现起来要考虑很多。
以下分别用排序数组和大小堆来实现。
使用大小堆:
import java.util.Arrays;
public class MedianInHeap {
/**
* 题目:设计方便提取中数的数据结构
* 设计一个数据结构,其中包含两个函数,1.插
- ajaxFileUpload 针对 ie jquery 1.7+不能使用问题修复版本
Chen.H
ajaxFileUploadie6ie7ie8ie9
jQuery.extend({
handleError: function( s, xhr, status, e ) {
// If a local callback was specified, fire it
if ( s.error ) {
s.error.call( s.context || s, xhr, status, e );
}
- [机器人制造原则]机器人的电池和存储器必须可以替换
comsci
制造
机器人的身体随时随地可能被外来力量所破坏,但是如果机器人的存储器和电池可以更换,那么这个机器人的思维和记忆力就可以保存下来,即使身体受到伤害,在把存储器取下来安装到一个新的身体上之后,原有的性格和能力都可以继续维持.....
另外,如果一
- Oracle Multitable INSERT 的用法
daizj
oracle
转载Oracle笔记-Multitable INSERT 的用法
http://blog.chinaunix.net/uid-8504518-id-3310531.html
一、Insert基础用法
语法:
Insert Into 表名 (字段1,字段2,字段3...)
Values (值1,
- 专访黑客历史学家George Dyson
datamachine
on
20世纪最具威力的两项发明——核弹和计算机出自同一时代、同一群年青人。可是,与大名鼎鼎的曼哈顿计划(第二次世界大战中美国原子弹研究计划)相 比,计算机的起源显得默默无闻。出身计算机世家的历史学家George Dyson在其新书《图灵大教堂》(Turing’s Cathedral)中讲述了阿兰·图灵、约翰·冯·诺依曼等一帮子天才小子创造计算机及预见计算机未来
- 小学6年级英语单词背诵第一课
dcj3sjt126com
englishword
always 总是
rice 水稻,米饭
before 在...之前
live 生活,居住
usual 通常的
early 早的
begin 开始
month 月份
year 年
last 最后的
east 东方的
high 高的
far 远的
window 窗户
world 世界
than 比...更
- 在线IT教育和在线IT高端教育
dcj3sjt126com
教育
codecademy
http://www.codecademy.com codeschool
https://www.codeschool.com teamtreehouse
http://teamtreehouse.com lynda
http://www.lynda.com/ Coursera
https://www.coursera.
- Struts2 xml校验框架所定义的校验文件
蕃薯耀
Struts2 xml校验Struts2 xml校验框架Struts2校验
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
蕃薯耀 2015年7月11日 15:54:59 星期六
http://fa
- mac下安装rar和unrar命令
hanqunfeng
mac
1.下载:http://www.rarlab.com/download.htm 选择
RAR 5.21 for Mac OS X 2.解压下载后的文件 tar -zxvf rarosx-5.2.1.tar 3.cd rar sudo install -c -o $USER unrar /bin #输入当前用户登录密码 sudo install -c -o $USER rar
- 三种将list转换为map的方法
jackyrong
list
在本文中,介绍三种将list转换为map的方法:
1) 传统方法
假设有某个类如下
class Movie {
private Integer rank;
private String description;
public Movie(Integer rank, String des
- 年轻程序员需要学习的5大经验
lampcy
工作PHP程序员
在过去的7年半时间里,我带过的软件实习生超过一打,也看到过数以百计的学生和毕业生的档案。我发现很多事情他们都需要学习。或许你会说,我说的不就是某种特定的技术、算法、数学,或者其他特定形式的知识吗?没错,这的确是需要学习的,但却并不是最重要的事情。他们需要学习的最重要的东西是“自我规范”。这些规范就是:尽可能地写出最简洁的代码;如果代码后期会因为改动而变得凌乱不堪就得重构;尽量删除没用的代码,并添加
- 评“女孩遭野蛮引产致终身不育 60万赔偿款1分未得”医腐深入骨髓
nannan408
先来看南方网的一则报道:
再正常不过的结婚、生子,对于29岁的郑畅来说,却是一个永远也无法实现的梦想。从2010年到2015年,从24岁到29岁,一张张新旧不一的诊断书记录了她病情的同时,也清晰地记下了她人生的悲哀。
粗暴手术让人发寒
2010年7月,在酒店做服务员的郑畅发现自己怀孕了,可男朋友却联系不上。在没有和家人商量的情况下,她决定堕胎。
12月5日,
- 使用jQuery为input输入框绑定回车键事件 VS 为a标签绑定click事件
Everyday都不同
jspinput回车键绑定clickenter
假设如题所示的事件为同一个,必须先把该js函数抽离出来,该函数定义了监听的处理:
function search() {
//监听函数略......
}
为input框绑定回车事件,当用户在文本框中输入搜索关键字时,按回车键,即可触发search():
//回车绑定
$(".search").keydown(fun
- EXT学习记录
tntxia
ext
1. 准备
(1) 官网:http://www.sencha.com/
里面有源代码和API文档下载。
EXT的域名已经从www.extjs.com改成了www.sencha.com ,但extjs这个域名会自动转到sencha上。
(2)帮助文档:
想要查看EXT的官方文档的话,可以去这里h
- mybatis3的mapper文件报Referenced file contains errors
xingguangsixian
mybatis
最近使用mybatis.3.1.0时无意中碰到一个问题:
The errors below were detected when validating the file "mybatis-3-mapper.dtd" via the file "account-mapper.xml". In most cases these errors can be d