生物信息学---蛋白质组学中氨基酸信息编码方式
特征编码:
1. 基于序列的特征:
资料来源:
蛋白质序列特征提取方法之——CKSAAP
1.1 CKSAPP(k空间氨基酸对的组成):
在CKSAAP(Compositon of k-spaced Amino Acid Pairs)方法中,利用在蛋白质序列片断中k个间隔距离的残基对(residue pairs)在该序列中的组成比例,建立数学模型,提取出特征向量,从而达到预测泛素(Ubiquitin)的目的。
残基:
组成多肽的氨基酸在相互结合时,由于其部分基团参与了肽键的形成而失去一分子水,因此把多肽中的氨基酸单位称为氨基酸残基。即由肽键连接的氨基酸失水后剩余部分。
泛素:
泛素(ubiquitin)是一种存在于所有真核生物(大部分真核细胞)中的小蛋白。 泛素由76个氨基酸组成,分子量大约8.451kDa。它的主要功能是标记需要分解掉的蛋白质,使其被26S蛋白酶体降解。26S蛋白酶体上调节亚基上的一些受体可以识别K48和K11位连接的多聚泛素化蛋白,20S核心亚基在ATPase供能下水解底物。泛素也可以标记跨膜蛋白,参与蛋白质的膜泡运输。非典型泛素链在细胞信号传导,内吞,以及DNA损伤修复,调控NF-κB通路中起着重要作用。它在真核生物中具有高度保守性,人类和酵母的泛素有96%的相似性。
数学计算的过程:
以长度为48的序列LEEYRKHVAERAAEGIAPKPLDANQMAALVELLKNPPAGEEEFLLDLL为例(编号是常见的20种氨基酸所代表的符号),
当k=0时,我们需要提取的残基对(residue pairs)为{LE,EE,EY,……,LD,DL,LL},即每个氨基酸和它相邻的下一个氨基酸组成一对提取出来,也就是说这两个氨基酸中间的间隔距离是k=0个氨基酸。以此类推,
当k=1时,我们需要提取的残基对为{LE,EY,ER,YK,……,LD,LL,DL}……本方法中,每一个氨基酸对都需要间隔一个氨基酸。
k最大为5。一共有6种情况(0,1,2,3,4,5)
细心的你一定会发现k=0时候,结尾会有1个氨基酸L没有配对,而提取的残基对数量为47;k=1时,有2个氨基酸LL没有配对,而提取的残基对数量为46;所以,规律就是,当序列长度为N,间隔为k时,一共可以提取的残基对数量为N-k-1,记为
NTotal=N−k−1
由于基本氨基酸数量为20,故而可以形成的残基对数量是20×20=400.我们统计的是这些残基对在这个蛋白质序列当中出现的概率,于是便产生了一个400维的特征向量,即

其中 NTotal 是总组成残基的长度(例如,如果长度为 L 的蛋白质片段残基为 31 且 k = 0、1、2、3、4 和 5,则NTotal = L - k - 1 将是分别为 30、29、28、27、26 和 25)。NAA、NAC、NAD、···、NYY代表片段内氨基酸对的频率。考虑到在本研究中执行 CKSAAP 方案k = 0 , 1 , 2 , 3 , 4 和 5 ,基于 CKSAAP 的特征向量的总维数为 400 × 6 = 2400。
1.2 PWAA(位置权重氨基酸)组成:
为了避免迷失序列顺序信息, Shi 等人[45]提出位置权重氨基酸组成 (position
weight amino acids composition, PWAA) 以提取蛋白质的序列信息。在许多蛋白质预
测的位点中都使用了这种方法,该方法能有效地提取目标位置附近的残差位置信息,
从而提高目标的精度。
给定一个氨基酸残基 ai(i = 1, 2,···, 20),ai在2L+1个氨基酸的序列片段P中的位置信息可以通过下式计算

其中 L 表示蛋白质序列片段 P 中来自中心位点的上游残基或下游残基的数量,如果ai是蛋白质序列片段 P 中的第 j 个位置残基,则 xi,j = 1,否则 xi,j = 0。通常情况下,残留物ai越靠近中心位置,Ci的绝对值越小。最后,基于 PWAA 的特征向量的维数为 20。
1.3 AARPC(氨基酸相对位置组成):
作者直接为每个氨基酸残基分配不同的位置整数值并计算氨基酸之间的相对位置。
- 使用从 1 到 20 的数字来编码 20 个氨基酸;
- 给定一个长度为 2L + 1 的蛋白质片段,每个残基距中心的位置记为 d,范围为 [-L, L];
- 对于蛋白质片段上的某个残基,对应的特征向量可以用nd来表示。最后,基于 AARPC 的特征向量的维数为 51。
1.4 氨基酸组成:
(1)氨基酸组成:(AAC)

(2)增强氨基酸:(EAAC)

(3)增强分组氨基酸: (EGAAC)
1.5 二肽组成(DC):

2. 基于物理化学性质的特征:
2.1 AAindex(氨基酸物理化学性质):
是一个代表氨基酸及成对氨基酸各种理化和生化性质数值指数的数据库。 AAindex 由三个部分组成:AAindex1、AAindex2 和 AAindex3。
其中,AAindex1 记录了 20 种氨基酸的数值指数,包含 544 个氨基酸指数,每个条目包含收录号、指数的简 短描述、参考信息以及 20 种氨基酸的属性值。
AAindex2 包含 94 个氨基酸置换矩阵: 67 个对称矩阵和 27 个非对称矩阵。
AAindex3 是统计的蛋白质接触电位,包含 47 个氨 基酸接触电位矩阵:44 个对称矩阵和 3 个非对称矩阵。
2.2 CTD(组成、过渡、分布):
CTD方案根据极性、中性和疏水性将20个氨基酸分为三类。然后根据其所属的类别,每个氨基酸由 1、2 或 3 编码。
组成 描述了 20 种天然氨基酸的总体百分比组成,定义为:其中 ns是编码序列中 s 的数量,L 是蛋白质片段序列的长度。

过渡(T) 表征一种天然氨基酸类型后跟另一种类型的氨基酸的百分比频率,可以通过以下方式计算:

其中 nxy 是分别编码为“xy”和“yx”的二肽的数量。
分布 (D) 测量每种类型的 20 种天然氨基酸的第一个,25%、50%、75% 和 100% 的各自位置,描述符 Ei 定义为:
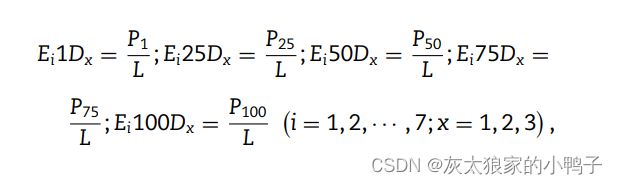
其中 P1、P25、P50、P75 和 P100 分别测量第一个残基的位置,x 的出现率分别为 25%、50%、75% 和 100%。最后,基于 CTD 的特征向量的维度是 (3 + 3 + 3 × 5) × 7 = 147。7是使用7种理化性质。
2.3 EBGW(基于分组权重的编码):


计算:
然后,根据以下不相交的组对氨基酸进行分配:C1 + C2 对 C3 + C4,或 C1 + C3 对 C2 + C4,或 C1 + C4 对 C2 + C3。对于蛋白质序列 P,它可以转化为三个二进制序列:

每一个二进制序列都可以分成一个长度增加的J个子序列。例如,对于 H1,第 j 个子序列定义为:

其中 Sum(j) 是第 j 个子序列中 1 的个数,D(j) = int ( j × L/J ) 指第j个子序列的长度,函数int()将小数四舍五入到最接近的整数。EBGW 方案将蛋白质序列定义为 3 × J 维向量。这里,J 被选择为 1、2、3、4 和 5。因此,基于 EBGW 的特征向量的维度为 3 × 15 = 45。
2.4 三联体组合信息编码(CT)
三联体组合信息编码 (cpnjoint triad, CT)[56]将氨基酸分成 7 类,每三个相邻的氨基酸为一个三联体,故有 7 x 7 x 7=343 种不同的三联体,每个三联体特征在蛋白质序
列中出现的频数 f i ( 1, 2,4... ,343) 为:对于每条蛋白质序列 P ,会得到 343 维特征向量。

3. 基于空间映射信息的特征:
先空着:
4. 进化信息:
4.1 BLOUSUM_62矩阵:
BLOSUM62 通过氨基酸置换矩阵来测量两条氨基酸段相似度,其反映了蛋白质进化信息, BLOSUM62 矩阵是在氨基酸序列的对比上,使两个肽序列的同一性不超过 62%,对 20 个氨基酸所获得的取代得分。对于序列长度为n 的蛋白质,最终形成一个n * M 维元素组成的矩阵, M 代表 20 种氨基酸。
5. 补充:
5.1 双剖面贝叶斯:(来源基于深度学习的蛋白质翻译后修饰位点预测相关问题研究_宋丽丽)



5.2 KNN算法:
K 近邻 (K-Nearest Neighbor, KNN) 算法[58]通过提取正、负样本相似序列中的特
征来获取修饰位点周围的局部序列相似性。两个查询序列片段c1和c2,序列c1和c2之
间的距离 Dist c c ( , ) 1 2 为: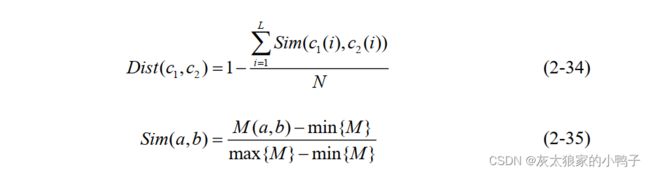
N 表示蛋白质序列窗口大小(氨基酸序列长度), Sim 是归一化的氨基酸替代矩阵, M 是
BLOSUM62 替换矩阵, a 和b 表示两个氨基酸, max/ min {M} 分别代表了替换矩阵
M 中的最大值与最小值。对于查询序列 p p p p ( , , , ) 1 2 L ,首先,计算 p 与相同数
量的正负样本之间的距离。其次,选出最近的k 个邻居。最后, k 个最近邻居中的正
邻居所占的百分比记作最终的 KNN 得分。