绿原酸酯与CALB Docking(3) - Covalent Docking
目录
- 绿原酸酯 与 calb 共价docking
-
- 目标
- 1. 结构准备
- 2. 结构预处理
- 3. Generate the alignment for ligand
- 4. 生成 PDBQT 文件
- 5. 生成 flexible/rigid PDBQT 文件
- 6. 生成 gpf 和 dpf 文件
- 7. AutoGrid and AutoDock
- 8. 查看结果
-
- HEE 与 绿原酸酯 结构对比
- 绿原酸酯在活性部位的两种可能取向
- 9. 如何去测试想法呢?
- cmd命令与操作
- 问题
-
- 1. protein.pdb文件中的待docking残基
- 2. ligand.pdb中待结合原子的定义和定位
- 3. 生成 ligcovalent.pdbqt 文件时的报错
- 4. 无法对蛋白生成 1lbs_protein_flex.pdbqt 文件
- 5. 多帧pdb与单帧pdb的区别
绿原酸酯 与 calb 共价docking
目标
1lbs的催化残基为Ser105。
- 将CALB 1lbs_protein.pdb 与 绿原酸丁酯分子 共价docking,比较与非共价docking(docking 1)的结果差异。
1. 结构准备
将 Docking(1)中的蛋白原始文件和配体pdb文件复制到新建的dock3文件夹中。
2. 结构预处理
处理1lbs.pdb文件,仅保留A链蛋白部分,得到1lbs_protein.pdb文件。
![]()
处理ligand.pdb文件,在酯键上构建C-O-C,形成以碳原子为中心的四面体结构。对结构进行简单优化。修改苯环上的键连关系为单-双键交替。得到ligand.mol2文件。
3. Generate the alignment for ligand
prepareCovalent.py 后,其结构如下:
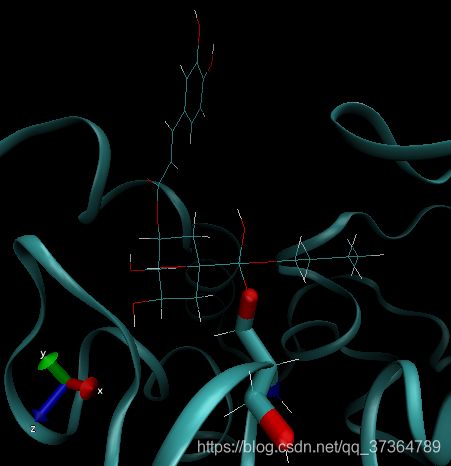
【注意到碳四面体上的羟基,带一个氢原子是否符合中间体(IS)的电荷分布?如何设置?(参考BB2010)】
4. 生成 PDBQT 文件
prepare_receptor4.py 后,得到 1lbs_protein.pdbqt 和 ligcovalent.pdbqt。
打开ligcovalent.pdbqt(备份一个文件) 和 ligcovalent.pdb 对比,修改原子电荷:
pdb:
HETATM 39 H SER A 105 56.929 2.738 33.839 1.00 0.00 H
pdbqt:
删去 39号氢原子;# HETATM 39 H SER A 105 56.929 2.738 33.839 1.00 0.00 0.215 HD
6号氧原子上加上氢原子的电荷,然后电荷减一 (-0.341 >> -1.126);# HETATM 6 OX SER A 105 57.380 2.354 34.607 1.00 0.00 -0.341 OA
5. 生成 flexible/rigid PDBQT 文件
得到 1lbs_protein_rigid.pdbqt 和 ligcovalent_flex.pdbqt 文件。
其中, 1lbs_protein_rigid.pdbqt 需要进行特殊处理,参见问题4。
6. 生成 gpf 和 dpf 文件
得到 1lbs_protein.gpf 和 ligcovalent_1lbs_protein.dpf。创建了空文件 empty。
7. AutoGrid and AutoDock
得到六个map文件,一个 maps.fld 和一个 maps.xyz
得到 ligcovalent_1lbs_protein.dlg 文件。
torsdof 17 # torsional degrees of freedom
Real= 11m 06.15s, CPU= 3m 52.88s, System= 0.45s
8. 查看结果
打开ADT。
载入1lbs_protein.pdbqt (1lbs_protein_rigid.pdbqt)文件,导出为protein.pdb文件。
载入ligcovalent_1lbs_protein.dlg,Build All,将所有的构象保存为empty_conf_0-9.pdb。
打开VMD。
载入protein.pdb文件。
载入empty_conf_0.pdb 文件。导入其余分子,保存为 empty_conf_all.pdb。
HEE 与 绿原酸酯 结构对比
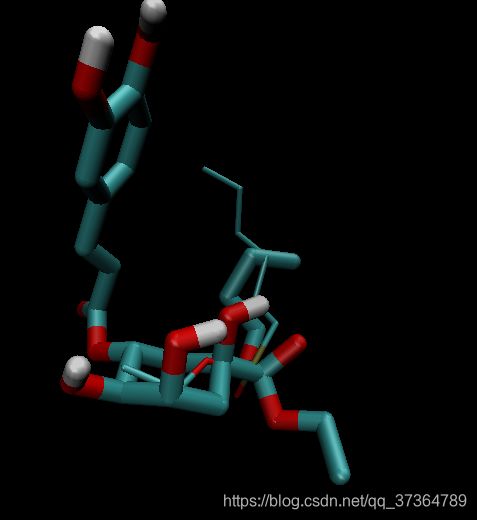
绿原酸酯在活性部位的两种可能取向
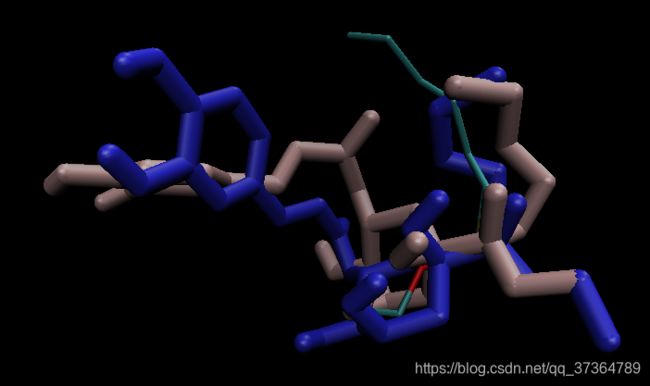
侧面来看,叠合所有的结构:
除了3和9之外的几个结构。
烷基链在下侧
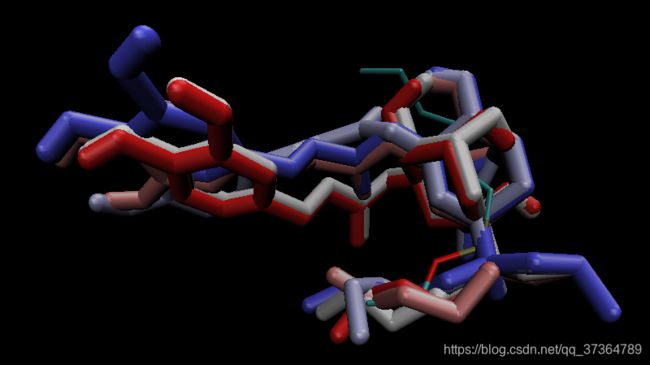
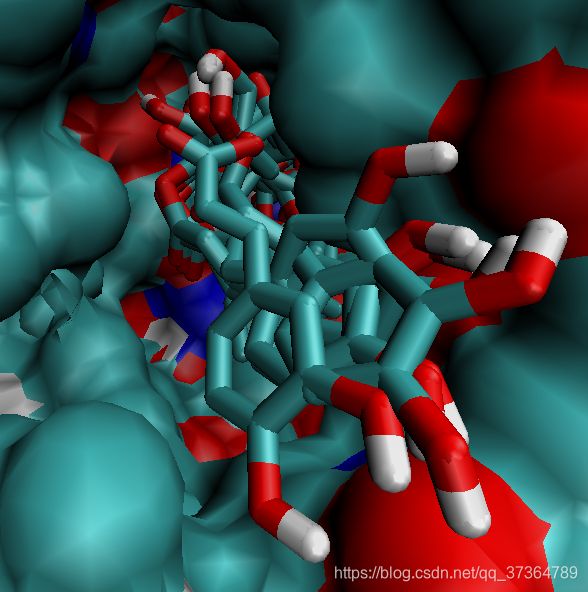
第3帧和第9帧(构象6和构象0.)
烷基链在上侧
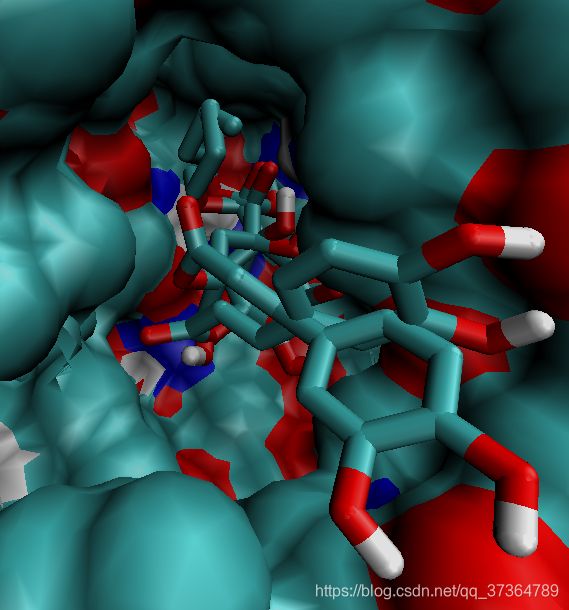
-
从传递角度来看,上面的口袋更加疏水,下面的口袋底部更加亲水。
-
以双分子反应的视角来看,第一种构象中,绿原酸与Ser105形成的共价Complex中,绿原酸配体完全堵塞了烷基链进入活性中心的道路。所以与之前的判断相同,第二种构象更可能是反应构象。
但是看起来反应道路也不太容易被堵塞;下述残基组成的疏水性通道可能使得烷基链从绿原酸分子下侧进入:

-
从稳定过渡态的角度来看,第一种构象中的O负离子对着氧负离子空洞,更有利于反应发生;第二类构象中实际发生了构象的反转。(但这可能是第一类构象出现次数较多的原因)
-
从精细调控的角度来看,远端对于苯环、酚的稳定似乎也值得考虑…
9. 如何去测试想法呢?
Rosseta 点突变?+ Redocking?
系统性的参数修改? >> 构象、评分变化?
Docking的原理进一步探究?
cmd命令与操作
# 1. 结构准备
cd D:\Desktop\adt
mkdir dock3
mkdir .\dock3\adcovalent
copy .\adCovalentDockResidue\adcovalent .\dock3\adcovalent /y
copy .\chloro\1lbs.pdb .\dock3\1lbs.pdb
copy .\chloro\ligand.pdb .\dock3\ligand.pdb
# 2. 结构预处理
# VS Code 对pdb文件进行操作
# GaussView 中 对 HEE 和绿原酸酯进行操作
# 3. Generate the alignment
cd dock3
"D:\Program Files (x86)\MGLTools-1.5.7\python.exe" ./adcovalent/prepareCovalent.py --ligand ./ligand.mol2 --ligindices 57,56 --receptor 1lbs_protein.pdb --residue A:SER105 --outputfile ligcovalent.pdb
# 4. 生成 PDBQT 文件
"D:\Program Files (x86)\MGLTools-1.5.7\python.exe" "D:\Program Files (x86)\MGLTools-1.5.7\Lib\site-packages\AutoDockTools\Utilities24\prepare_receptor4.py" -r 1lbs_protein.pdb -A hydrogens
"D:\Program Files (x86)\MGLTools-1.5.7\python.exe" "D:\Program Files (x86)\MGLTools-1.5.7\Lib\site-packages\AutoDockTools\Utilities24\prepare_receptor4.py" -r ligcovalent.pdb
# 修改ligcovalent.pdbqt中的电荷设置
# 5. 生成 flexible/rigid PDBQT 文件
# 对于 1lbs_protein 文件进行一定的预处理,以正确生成文件。
"D:\Program Files (x86)\MGLTools-1.5.7\python.exe" "D:\Program Files (x86)\MGLTools-1.5.7\Lib\site-packages\AutoDockTools\Utilities24\prepare_flexreceptor4.py" -r 1lbs_protein.pdbqt -s 1lbs_protein:A:SER105
"D:\Program Files (x86)\MGLTools-1.5.7\python.exe" "D:\Program Files (x86)\MGLTools-1.5.7\Lib\site-packages\AutoDockTools\Utilities24\prepare_flexreceptor4.py" -r ligcovalent.pdbqt -s ligcovalent:A:SER105
# 6. 生成 gpf 和 dpf 文件
"D:\Program Files (x86)\MGLTools-1.5.7\python.exe" "D:\Program Files (x86)\MGLTools-1.5.7\Lib\site-packages\AutoDockTools\Utilities24\prepare_gpf4.py" -r 1lbs_protein_rigid.pdbqt -x ligcovalent_flex.pdbqt -l ligcovalent_flex.pdbqt -y -I 20 -o 1lbs_protein.gpf
"D:\Program Files (x86)\MGLTools-1.5.7\python.exe" "D:\Program Files (x86)\MGLTools-1.5.7\Lib\site-packages\AutoDockTools\Utilities24\prepare_dpf4.py" -r 1lbs_protein_rigid.pdbqt -x ligcovalent_flex.pdbqt -l ligcovalent_flex.pdbqt -o ligcovalent_1lbs_protein.dpf -p move=empty
# 编辑 ligcovalent_1lbs_protein.dpf
# unbound_energy 0.0 # 修改命令 unbound_model bound >> unbound_energy 0.0
copy nul empty # 生成 empty 空文件
# 7. AutoGrid and AutoDock
D:\Desktop\adt\autogrid4.exe -p 1lbs_protein.gpf -l 1lbs_protein.glg
D:\Desktop\adt\autodock4.exe -p ligcovalent_1lbs_protein.dpf -l ligcovalent_1lbs_protein.dlg
# 修正 .dlg 文件中的问题 # '-1.#j' >> ' -nan'
# 8. 查看结果
问题
1. protein.pdb文件中的待docking残基
这是 3upo:B:222 Ser222的定义:
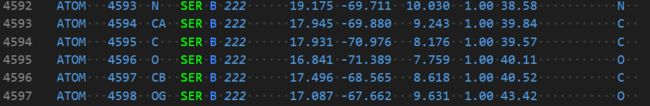
这是 1lbs:A:105 Ser222的定义:
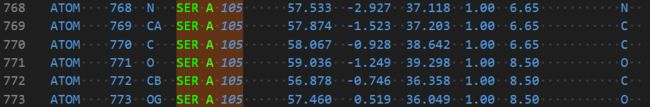
无需做修改。
2. ligand.pdb中待结合原子的定义和定位
penicillin G的原子定位,待结合的是C1 和 O2,docking命令为:
"D:\Program Files (x86)\MGLTools-1.5.7\python.exe" ./adcovalent/prepareCovalent.py --ligand ./3upo_test/ligand.mol2 --ligindices 1,2 --receptor ./3upo_test/3upo_protein.pdb --residue B:SER222 --outputfile ligcovalent.pdb

这是ligand.mol2的lables,将对接的原子是C55-O56-C57.
因此,命令为
"D:\Program Files (x86)\MGLTools-1.5.7\python.exe" ./adcovalent/prepareCovalent.py --ligand ./3upo_test/ligand.mol2 --ligindices 57,56 --receptor ./3upo_test/3upo_protein.pdb --residue B:SER222 --outputfile ligcovalent.pdb
3. 生成 ligcovalent.pdbqt 文件时的报错
报错的电荷设置如下:
D:\Desktop\adt\dock3>“D:\Program Files (x86)\MGLTools-1.5.7\python.exe” “D:\Program Files (x86)\MGLTools-1.5.7\Lib\site-packages\AutoDockTools\Utilities24\prepare_receptor4.py” -r ligcovalent.pdb
swig/python detected a memory leak of type ‘BHtree *’, no destructor found.
adding gasteiger charges to peptide
Sorry, there are no Gasteiger parameters available for atom ligcovalent:A:SER105:OX
Sorry, there are no Gasteiger parameters available for atom ligcovalent:A:SER105:OX
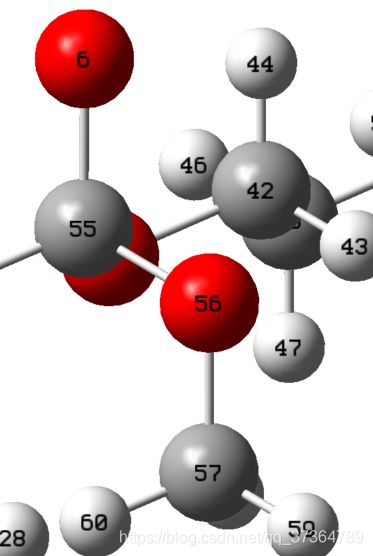
打开ligcovalent.pdbqt文件,发现O8和O9上没有正确的电荷,这是两个酚基氧原子。
仔细看了一下ligand.mol2文件和ligcovalent.pdb文件,由于键连关系判断错误;多加了一些错误的氢原子;导致生成的pdb文件不正确。
如下图,19-24的苯环目前成键关系是混乱的。

将键连关系改为交替的单双键。
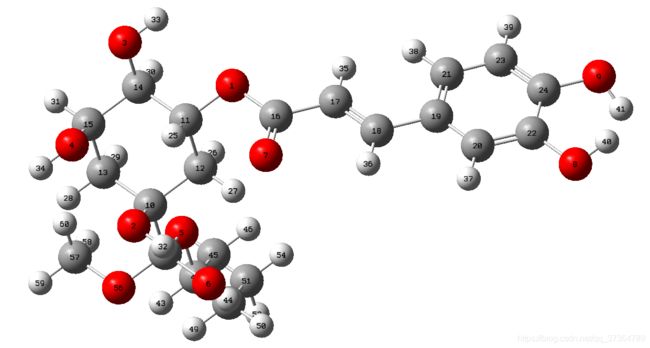
重新生成ligcovalent.pdb文件。
重新生成ligcovalent.pdbqt文件。
4. 无法对蛋白生成 1lbs_protein_flex.pdbqt 文件
"D:\Program Files (x86)\MGLTools-1.5.7\python.exe" "D:\Program Files (x86)\MGLTools-1.5.7\Lib\site-packages\AutoDockTools\Utilities24\prepare_flexreceptor4.py" -r 1lbs_protein.pdbqt -s 1lbs_protein:A:SER105
与在测试案例中类似,上述语句无法正常生成所要的_rigid和_flex文件。
ligcovalent.pdbqt与1lbs_protein.pdbqt的区别在于文件大小不同,怀疑与文件大小有关。
测试一系列的test.pdbqt文件。
"D:\Program Files (x86)\MGLTools-1.5.7\python.exe" "D:\Program Files (x86)\MGLTools-1.5.7\Lib\site-packages\AutoDockTools\Utilities24\prepare_flexreceptor4.py" -r test.pdbqt -s test:A:SER105
154 KB 不可;跑一会儿之后不生成东西。
114 KB可以:跑一下之后得到结果。【奇奇怪怪】
75 KB可以:跑一下之后得到结果。
根据报错信息和这个大小的测试结果,也很难说如何去修正问题(修改缓存?)。一个补丁的做法是先手动修改.pdbqt 文件。再拖一拖看看如何改这个问题…
目测其效果就是把 Ser105 的CA, CB, OG, HG 四个原子摘出去。保留其余部分。
test.pdbtest_rigid.pdb
test_flex.pdb


用手动补丁方式,把 test_rigid.pdbqt 的后面的内容从1lbs_protein.pdbqt复制过来。修改文件名为1lbs_protein_rigid.pdbqt
出现一个原子序数的断层。
5. 多帧pdb与单帧pdb的区别
多帧(frame) pdb 之间以 END 分隔。

按照 帧数着色:Coloring Method -> Trajectory -> Timestep