R语言——taxize(第一部分)
ropensci 系列之 taxize (中译手册)
- taxize 包
-
- 1. taxize支持的网络数据源
-
- 简介
- 目前支持的API:
- 针对Catalogue of Life(COL)
- 2. 浅尝 taxize 的一些使用例子
-
- 2.1. **从NCBI上获取唯一的分类标识符**
- 2.2. **获取分类信息**
- 2.3. 获得类群的直系子类群
- 2.4. 向下检索子类群直至某个阶元
- 2.5. 向上检索类群直至某个阶元
- 2.6. 检索同物异名
- 2.7. 从多个数据源获得分类标识符
- 2.8. 根据科学名检索俗名
- 2.9. 根据俗名检索科学名
- 2.10. 检索多个类群的最近共有父类群
- 2.11. 强制转换
- 3. taxize 文档中译
-
- 3.1. apg(检索APG系统中的名称)
- 3.2. apg_families(MOBOT的科名,现成的数据)
- 3.3. apg_orders(MOBOT的目名)
- 3.4. apg_lookup(在APGⅢ的类群名称查找并替换名称)
- 3.5. bold_downstream(在BOLD的阶元结构中向下检索所有类群名称)
- 3.6. bold_search(根据分类标识符查找生命条形码)
- 3.7. children(根据给定的类群名称或ID检索直接子类群)
- 3.8. class2tree(将分类信息列表转换为树状)
taxize 包
- 标题:来自网络的分类信息
- 说明:与一系列网络 APIs进行交互,完成例如获取数据库特定的分类标识符,验证物种名称,获取分类层次结构,获取下游和上游分类名称,获取分类同义词,将科学名称转换为常用名称,反之亦然,等等。
1. taxize支持的网络数据源
来自网络的分类信息。
简介
+ 允许用户在许多网站上搜索物种名称(科学的和常见的),下载上下游的分类等级信息,以及许多其他东西。
+ 包中特定API的函数有一个由下划线分隔的前缀和后缀。它们遵循service_whatitdoes的格式。例如,gnr_resolve使用Global Names Resolver的API来解析物种名称。
+ 包中不涉及特定API的通用函数没有下划线分隔的两个单词,例如classification。
+ 某些数据源需要API密钥。有关更多信息,请参阅taxize -authentication。
目前支持的API:
| 中译名 | API | 前缀 | SOAP? |
|---|---|---|---|
| 网络生命大百科 | Encyclopedia of Life(EOL) | eol | FALSE |
| 综合分类信息服务 | Integrated Taxonomic Information Service(ITIS) | itis | FALSE |
| 全球名称解析器 | Global Names Resolver(from EOL/GBIF) | gnr | FALSE |
| 全球名称名录 | Global Names Index(from EOL/GBIF) | gni | FALSE |
| 国际自然保护联盟濒危物种红色名录 | IUCN Red List | iucn | FLASE |
| Tropicos数据库 | Tropicos(from Missouri Botanical Garden) | tp | FALSE |
| 植物名录 | Theplantlist.org | tpl | FLASE |
| 国家生物技术信息中心 | National Center for Biotechnology Information | ncbi | FALSE |
| 加拿大维管植物数据库 | CANADENSYS Vascan name search API | vascan | FALSE |
| 国际植物名称名录 | International Plant Names Index(IFNI) | ipni | FALSE |
| 世界海洋物种名录 | World Register of Marine Species(WoRMS) | worms | TRUE |
| 生命条形码数据库 | Barcode of Life Data Systems(BOLD) | bold | FALSE |
| 泛欧物种名录基础 | Pan-European Species directories Infrastructure(PESI) | pesi | TRUE |
| 真菌数据库 | Mycobank | myco | TRUE |
| 生物多样性网络 | National Biodiversity Network(UK) | nbn | FALSE |
| 全球真菌名录 | Index Fungorum | fg | FALSE |
| 欧洲多样性观察网 | EU BON | eubon | FALSE |
| 世界名称名录 | Index of Names(ION) | ion | FALSE |
| 生命之树 | Open Tree of Life(TOL) | tol | FALSE |
| 北美自然保护区 | NatureServe | natserv | FALSE |
如果上面的源在SOAP?列中显示TRUE ,则该资源在此包中不可用。它们可以从另一个名为 taxizesoap 的软件包中获得。请参阅GitHub repo了解如何安装https://github.com/ropensci/taxizesoap
针对Catalogue of Life(COL)
COL最近在2019年引入了速率限制-这使得API基本上无法使用——COL +即将推出,当它稳定时我们将在这里合并它。参见https://github.com/ropensci/colpluz获取CoL+的R实现。
2. 浅尝 taxize 的一些使用例子
目前尚未找到完备的关于taxize的用户手册,暂且以https://www.rdocumentation.org/packages/taxize/versions/0.9.4中的内容为引,先体验一下taxize。
大部分“taxize”都围绕分类标识符展开。众所周知,分类名称是很混乱的,比如拼写错误,同物异名等等。先获取能让数据源识别的标识符(ID),然后才能继续获取其他分类信息。
网页给出了下面的例子:
2.1. 从NCBI上获取唯一的分类标识符
uids <- get_uid(c("Chironomus riparius", "Chaetopteryx"))
以及运行后在控制台的输出,可以看到输出结果还是非常详细的,包括请求数量、请求内容、请求结果以及汇总报告。
No ENTREZ API key provided
Get one via taxize::use_entrez()
See https://ncbiinsights.ncbi.nlm.nih.gov/2017/11/02/new-api-keys-for-the-e-utilities/
══ 2 queries ═══════════════
Retrieving data for taxon 'Chironomus riparius'
✔ Found: Chironomus+riparius
Retrieving data for taxon 'Chaetopteryx'
✔ Found: Chaetopteryx
══ Results ═════════════════
• Total: 2
• Found: 2
• Not Found: 0
而且,返回的结果也不单单是标识符。尝试输出uids后,可以看到该变量里保存的信息非常有用且简洁。
以下是输入:
uids
以下是输出:
[1] "315576" "492549"
attr(,"class")
[1] "uid"
attr(,"match")
[1] "found" "found"
attr(,"multiple_matches")
[1] FALSE FALSE
attr(,"pattern_match")
[1] FALSE FALSE
attr(,"uri")
[1] "https://www.ncbi.nlm.nih.gov/taxonomy/315576"
[2] "https://www.ncbi.nlm.nih.gov/taxonomy/492549"
通过查看get_uid()的帮助页面,可以了解到其他属性的含义。match表示查找结果如何,mutiple_matches表示是否查找到多项匹配结果,pattern_match表示当存在多项匹配时,是否生成了最佳匹配结果,uri表示在此链接有关于当前类群更多的信息。
2.2. 获取分类信息
什么是分类信息?比如说现在有一个物种,然后分类信息就是从种级向上的所有分类等级,包括属、科、目、纲、界。
通过第一步获得的标识符查看它们的分类信息:
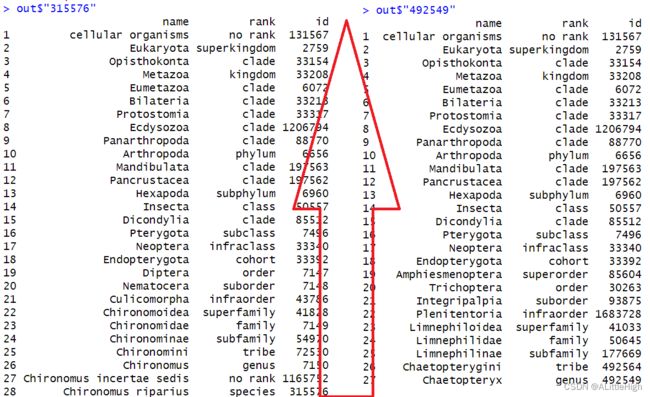
out <- classification(uids)
结果是数据框列表

细心一点可以发现,这两个类群的分类阶元至少有27层,这是因为除了界门纲目科属种外,许多中间阶元,例如亚属、亚科、超科、超目等都会包含在分类信息中。
在分别查看这两个数据框时,注意标识符要用字符。分类阶元从上到下逐级减小,直至我们输入的标识符对应的类群。
2.3. 获得类群的直系子类群
这个方法并不需要提前获取类群的标识符,也不需要声明类群的分类阶元。
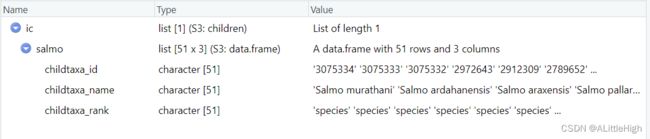
让我们先尝试一下获取属级类群的直系子类群,也就是属下的所有物种,以Salmo为例。
ic <- children("salmo", db ="ncbi")
注意直系子类群有很强的限制性,一般来说,标准的分类阶元是界门纲目科属种,但是正如前文获取分类信息时一样,实际上还会包含超目、亚科等阶元。所以在使用时要返回的结果可能与期望大相径庭,如果要查看某个科中的属级类群有哪些?
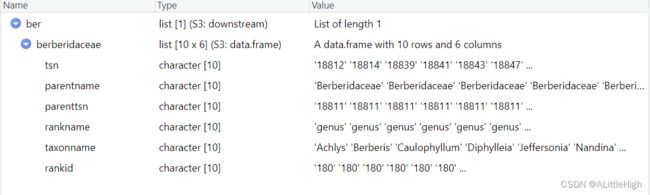
ber <- children("berberidaceae", db="ncbi")
实际上,返回的都是亚科类群。

2.4. 向下检索子类群直至某个阶元
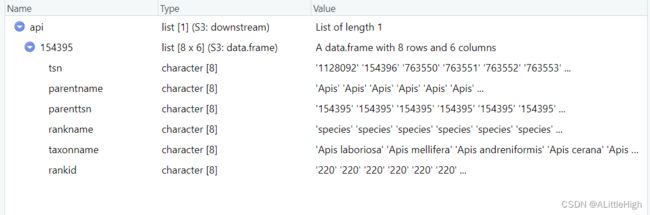
从字面上看,此方法可以实现多层阶元检索子类群。我们先过一下给出的例子:获得Apis的所有物种。
api <- downstream(as.tsn(154395), db = 'itis', downto = 'species', verbose = FALSE)
再多尝试一次,依旧以berberidaceae为例,查找该科下的所有属。
ber <- downstream("berberidaceae", db="itis", downto="genus", verbose=FALSE)
══ 1 queries ═══════════════
Retrieving data for taxon 'berberidaceae'
✔ Found: berberidaceae
══ Results ═════════════════
• Total: 1
• Found: 1
• Not Found: 0
返回的结果中包含了所有berberidaceae的属,只不过没有中间阶元,比如亚科。
2.5. 向上检索类群直至某个阶元
从物种Pinus contorta向上检索至属级阶元,具体效果如何见下:
pi <- upstream("Pinus contorta", db = 'itis', upto = 'Genus', verbose=FALSE)
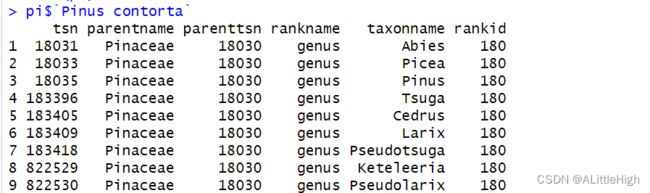
实际上可以这样理解,该方法从目标物种向上级检索到直接父类群,再找到此父类群的所有姊妹类群(同阶元类群)。
2.6. 检索同物异名
这个就很好理解了,直接看看例子:
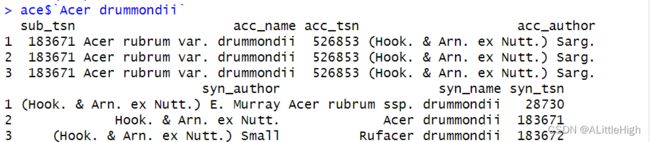
ace <- synonyms("Acer drummondii", db="itis")
*在
══ 1 queries ═══════════════
Retrieving data for taxon 'Acer drummondii'
✔ Found: Acer drummondii
══ Results ═════════════════
• Total: 1
• Found: 1
• Not Found: 0
Accepted name(s) is/are 'Acer rubrum var. drummondii'
Using tsn(s) 526853
2.7. 从多个数据源获得分类标识符
本例子非常清晰明了,当然我们在这里不会详细介绍使用的方法的参数。
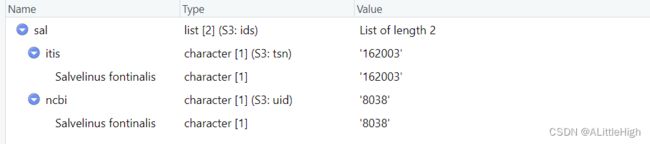
sal <- get_ids(sci_com ="Salvelinus fontinalis", db = c('itis', 'ncbi'), verbose=FALSE)
══ db: itis ═════════════════
══ 1 queries ═══════════════
Retrieving data for taxon 'Salvelinus fontinalis'
✔ Found: Salvelinus fontinalis
══ Results ═════════════════
• Total: 1
• Found: 1
• Not Found: 0
══ db: ncbi ═════════════════
No ENTREZ API key provided
Get one via taxize::use_entrez()
See https://ncbiinsights.ncbi.nlm.nih.gov/2017/11/02/new-api-keys-for-the-e-utilities/
══ 1 queries ═══════════════
Retrieving data for taxon 'Salvelinus fontinalis'
✔ Found: Salvelinus+fontinalis
══ Results ═════════════════
• Total: 1
• Found: 1
• Not Found: 0
2.8. 根据科学名检索俗名
ha <- sci2comm('Helianthus annuus', db = 'itis')
[1] "common sunflower" "sunflower" "wild sunflower" "annual sunflower"
2.9. 根据俗名检索科学名
bb <- comm2sci("black bear", db = "itis")
[1] "Ursus americanus luteolus" "Ursus americanus" "Ursus americanus"
[4] "Ursus americanus americanus" "Chiropotes satanas" "Ursus thibetanus"
[7] "Ursus thibetanus"
2.10. 检索多个类群的最近共有父类群
lc <- lowest_common(c("Sus scrofa", "Homo sapiens", "Nycticebus coucang"), db = "ncbi")
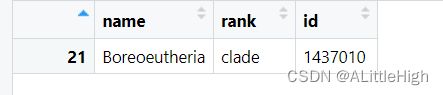
也许你注意到了行号是21,这可能意味着lowest_common方法是先找到所有类群的所有分类信息,即classification,然后交叉比较后抽取了某行分类信息。
2.11. 强制转换
- 从数值型至uid类型:as.uid(315567)
- 从列表型至uid类型:as.uid(list(“315567”,“3339”))
- 从uid类型至数据框类型:data.frame(as.uid(c(315567, 3339)))
3. taxize 文档中译
资源https://cran.r-project.org/web/packages/taxize/taxize.pdf
介绍函数方法时,并不严格遵循文档的顺序。
3.1. apg(检索APG系统中的名称)
描述:被子植物分类系统,又称APGⅢ,中的类群名称和替代名。
用法:apgOrders(…),apgFamilies(…)
参数:…(传递给crul::verb-GET)
参考:http://www.mobot.org/MOBOT/research/APweb/
示例:结果参考 3.2. apg_families 和 3.3. apg_orders。
3.2. apg_families(MOBOT的科名,现成的数据)
描述:APGⅢ中的科名和替代名。
格式:该数据框有1705行,6列。
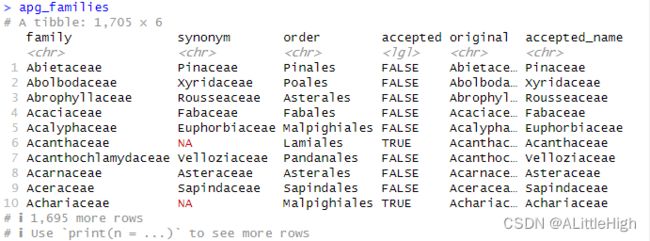
family:科名。
synonym:如果accepted列为FALSE,此项就是接受名;如果accepted列为TRUE,此项为NA。
order:目名。
accepted:逻辑值,取决于family列的名称是否被接受。
original:APG网站的原始数据。当此项存在时,family列的名称将会映射到此项。
accepted_name:接受名。整合family和synonym列后的结果。
说明:本数据集是在2020-06-03借助 apgFamilies() 生成的。
3.3. apg_orders(MOBOT的目名)
描述:APGⅢ中的科名和替代名。
格式:该数据框有576行,5列。
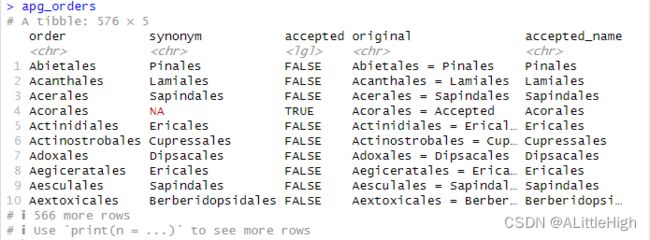
order:目名。
synonym:如果accepted列为FALSE,此项就是接受名;如果accepted列为TRUE,此项为NA,那 么order列为接受名。
accepted:逻辑值,取决于order列是否为接受名。
original:APG网站的原始数据。当此项存在时,order列的名称将会映射到此项。
accepted_name:接受名。整合order和synonym列后的结果。
说明:本数据集是在2020-06-03借助 apgOrders() 生成的。
3.4. apg_lookup(在APGⅢ的类群名称查找并替换名称)
描述:在APGⅢ的类群名称查找并替换名称。
用法:apg_lookup(taxa, rank=“family”)。
参数:
- taxa:(字符型)在APGⅢ中查找该分类名称的替换名。
- rank:(字符型)分类阶元,接受值为family或order。
说明:本方法在内部调用的是 apg_families 和 apg_orders 这两个数据集。
返回值:APGⅢ中的科名或目名,如果参数taxa的名称与APG中的相同,返回原始名称,否则返回NA。
示例:
-
发现新名称
apg_lookup(taxa = "Hyacinthaceae", rank = "family")new name... [1] "Asparagaceae" -
相同名称
apg_lookup(taxa = "Poaceae", rank = "family")name is the same... [1] "Poaceae" -
检索失败
apg_lookup(taxa = "Foobar", rank = "family")no match found... [1] NA
3.5. bold_downstream(在BOLD的阶元结构中向下检索所有类群名称)
用法:bold_downstream(id, downto, intermediate = FALSE, …)
参数:
- id:(整数值)单个或多个BOLD的分类标识符。
- downto:(字符值)期望向下到达的分类阶元。分类阶元区分大小写!,详见data(rank_ref)。
- intermediate:(逻辑值)TRUE时,返回包含期望阶元的类群名称和一个包含中间阶元类群数据框的列表的列表。默认为FALSE。
- …:传递给crul::verb-GET。
说明:此方法需要爬取BOLD网站,故而不太稳定。
返回值:当intermediate为FALSE时,返回的是储存期望阶元的分类信息的数据框;而当intermediate为TRUE时,则返回一个列表,列表长度为2,包括期望阶元的类群名称和中间阶元的名称。
示例:
-
intermediate = FALSE,即默认
bold_downstream(id = 3451, downto = "species")name id rank 1 Gadus chalcogrammus 360473 species 2 Gadus macrocephalus 19837 species 3 Gadus morhua 26136 species 4 Gadus ogac 747382 species 5 Gadus sp. 674263 species 6 Gadus sp. OPC-2017 794750 species -
intermediate = TRUE
i <- bold_downstream(id = 443, downto = "genus", intermediate = TRUE)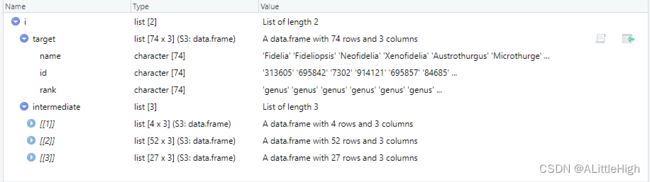
3.6. bold_search(根据分类标识符查找生命条形码)
用法:bold_search(sci = NULL, id =NULL, fuzzy = FALSE, dataTypes = “basic”, includeTree = FALSE, response = FALSE, name = NULL, …)
参数:
- sci:(字符型)单个或多个科学名。
- id:(整数型)单个或多个BOLD分类标识符。
- fuzzy:(逻辑值)决定是否采用模糊检索,默认FALSE。只有使用了sci参数时才有用。
- dataTypes:(字符型)决定返回的数据类型,详见说明。当使用了sci参数时会忽略此参数,当使用id参数时才有用。
- includeTree:(逻辑值)默认FALSE,如果为TRUE,返回一个包含父类群和期望类群的信息的列表,只有使用了id参数才有用。
- response:(逻辑值)此参数是返回的curl响应,对于调试很有用,也能获得API响应的详细信息。
- name:已弃用!,请使用sci。
- …:传递给crul::verb-GET。
说明:必须向此方法传递sci或id参数之一,其余参数都是可选的。dataTypes参数的接受值:
| 接受值 | 返回说明 |
|---|---|
| all | 所有数据 |
| basic | 基本的类群信息 |
| images | 标本图片。包含版权信息,图片链接,图片元数据 |
| stats | 标本和测序统计。包括公开物种数,公开BIN数,公开标记物数,公开记录数,标本数,测序物种数,条形码标本数,物种数,条形码物种数 |
| geo | 采集点信息。包括国家和采集点地图 |
| sequencinglabs | 测序实验室。包括实验室名称和记录数 |
| depository | 标本仓库。包括仓库名和记录数 |
| thirdparty | 第三方信息。包括维基百科总结,维基百科链接和GBIF地图 |
返回值:一个由数据框组成的列表。
参考:http://www.boldsystems.org/index.php/resources/api
示例:
-
简单的例子
bold_search(sci="Apis")input taxid taxon tax_rank tax_division parentid parentname taxonrep specimenrecords representitive_image.image representitive_image.apectratio 1 Apis 1937 Apis genus Animalia 878935 Apini Apis 5222 BOFTH/B3260-B09+1247093258.jpg 1.362 -
模糊查找
a <- bold_search(sci="achl", fuzzy = TRUE)
-
当使用id,同时可以使用dataTypes和includeTree
bold_search(id=88899, dataTypes = "basic", includeTree = TRUE)input taxid taxon tax_rank tax_division parentid parentname taxonrep 1 88899 18 Chordata phylum Animalia 1Chordata 2 88899 51 Aves class Animalia 18 Chordata Aves 3 88899 339 Coraciiformes order Animalia 51 Aves 4 88899 88898 Momotidae family Animalia 339 Coraciiformes 5 88899 88899 Momotus genus Animalia 88898 Momotidae
3.7. children(根据给定的类群名称或ID检索直接子类群)
描述:本方法区别于 downstream() 的点在于,children()只搜集直接子类群,而downstream()是搜集期望子阶元的分类名称。
用法:
## Default S3 method:
children(...)
## S3 method for class 'tsn':
children(sci_id, db=NULL, rows=NA, x=NULL, ...)
## S3 method for class 'wormsid':
children(sci_id, db=NULL, ...)
## S3 method for class 'ids':
children(sci_id, db=NULL, ...)
## S3 method for class 'uid':
children(sci_id, db=NULL, ...)
## S3 method for class 'boldid':
children(sci_id, db=NULL, ...)
参数:
- …:其他额外传递给ritis::hierarchy_down(),ncbi_children(),worrms::wm_children(),bold_children()的参数。
- sci_id:类群名称(字符型)或ID(字符型或数值型)向量。
- db:(字符型)用于检索的数据源。接受值是itis,ncbi,worms或bold之一。每个数据源都有自己特有的一套标识符,不属于指定数据源的标识符也有可能返回检索结果,只不过是错误的。
- rows:(数值型)从1至无穷大的整数值。当为NA(默认),所有行都有效。注意,如果传入任何可接受类的分类id: tsn,则忽略此参数。NCBI有这个函数的方法,但是rows参数不起作用。
- x:已弃用,见 sci_id。
返回值:一个带命名的数据框,储存每个输入类群的所有子类群名称。如果在数据源中没有匹配项,则返回NA。
ncbi:当数据源是 ncbi 时,默认ambigous = TRUE,表示像“unclassified”、“unknown”、“uncultured”和“sp.”的子类群都不会舍弃。
bold:数据源bold存在连接不稳定的情况。
示例:
-
输入ID
children(161994, db = "itis")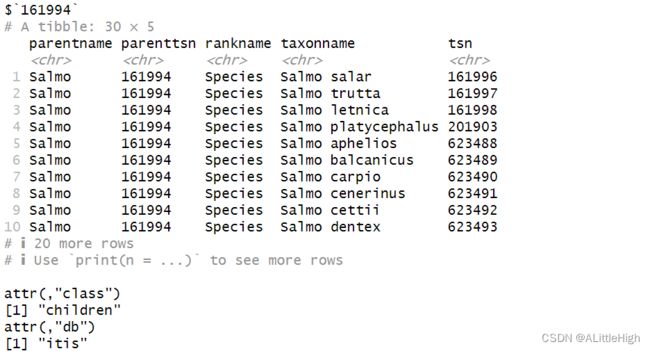
-
输入名称
children("Salmo", db = 'itis')
-
使用两个数据源
t <- children(get_ids("Apis", db = c('ncbi','itis')))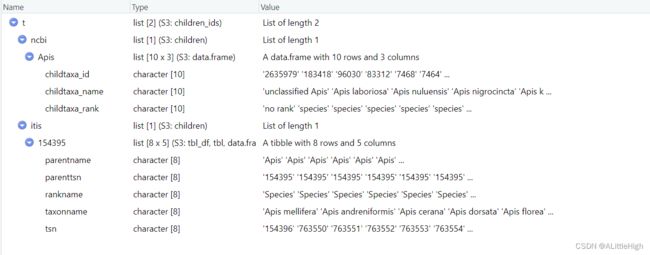
3.8. class2tree(将分类信息列表转换为树状)
描述:将多个独立物种的阶元层级列表以分类阶元矩阵的形式塞入一个物种内,然后仅根据分类计算距离矩阵,随后输出phylo或dist对象。
用法:
class2tree(input, varstep=TRUE, check=TRUE, ...)
## S3 method for class 'classtree'
plot(x, ...)
## S3 method for class 'classtree'
print(x, ...)
参数:
- input:classification()返回的数据框列表。
- varstep:根据不同类别数量的比例损失,改变连续阶元之间的步长。
- check:为TRUE时,删除所有行不同或行不变的冗余阶元,并将每行视为不同的基础分类类群(物种)。如果为FALSE,所有阶元都被保留,并且基础分类类群(物种)也必须编码为变量(列)。
- …:传递给hclust的参数。
- x:class2tree()返回的结果,用于输出或绘图。
说明:详见vegan::taxa2dist()。生成分类树不仅依赖分类阶元的聚类,还会使用实际的类群枝。本方法的流程如下:首先,从输入中搜集每个类群的可用分类阶元和对应的ID;然后整合所有类群的阶元向量,生成一个矩阵,矩阵的列是所有类群的有序分类阶元,行是这些类群的阶元向量;随后这个阶元矩阵转换为分类ID矩阵,缺失的阶元会由原阶元名称生成的伪ID代替;最后,ID矩阵用于将具有相似分类阶元层级的类群聚类。
返回值:一个带槽位的classtree对象。当直接输出返回值时,只会显示phylo部分,其余三部分可以通过output$classification查看。
- phylo:结果。
- classification:分类信息数据框,类群为行,分类信息阶元为列。
- distmat:距离矩阵。
- names:系统发育端点的名称。
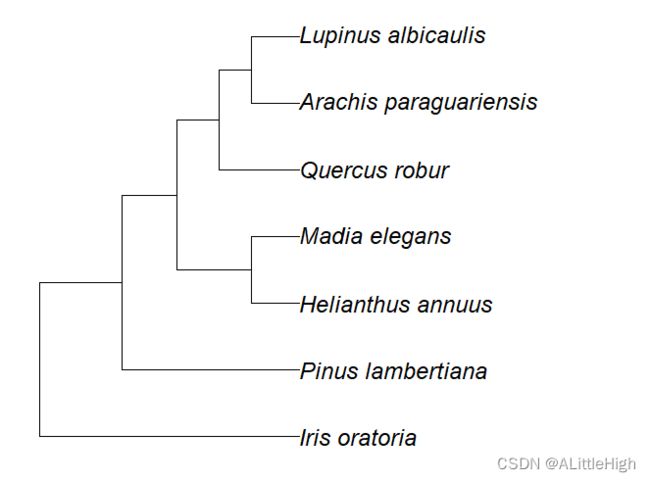
示例:
spnames <- c('Quercus robur', 'Iris oratoria', 'Arachis paraguariensis',
'Helianthus annuus','Madia elegans','Lupinus albicaulis',
'Pinus lambertiana')
out <- classification(spnames, db='itis')
tr <- class2tree(out)