基因组浏览器Jbrowse2使用和配置,支持多种格式的基因组文件
基因组浏览器Jbrowse2使用和配置,支持多种格式的基因组文件
-
- 1.准备工作
- 2.部署项目
- 3.启动项目
- 4.配置本地基因组信息
- 4.删除配置
- 5.配置默认展示
本文严禁转载!如需转载请注明来源!
本文链接:https://blog.csdn.net/Opticalproperti/article/details/130282800
1.准备工作
JBrowse 是一种新型的基因组浏览器,可以在网络上、桌面上或嵌入到您的应用程序中运行。
以下内容是介绍如何将 JBrowse2嵌入浏览器页面。
官网文档地址: JBrowse2官方文档
官方配置相关: JBrowse2官方文档配置
1.服务器:我尝试过了Ubuntu和CentOS8 两种服务器,虽然都可以直接运行,但是在使用过程中发现,官网的liunx代码都是Ubuntu系统的,所以当前建议使用Ubuntu服务器。
2.FASTA文件:创建 jbrowse 配置的第一步是加载基因组程序集。必须准备基因组相对应的 fa文件,该文件后缀名为 .fa。
2.部署项目
当前是根据官方文档:JBrowse web setup using the CLI 的方式进行项目的配置和启动。
1.需要nodeJs环境:在服务器上安装node环境,并且Jbrowse不支持低版本的node启动,建议直接去官网下载最近版本的nodejs。
2.安装Jbrowse CLI
npm install -g @jbrowse/cli //通过命令直接下载
下载完成后使用代码检查是否下载成功
jbrowse --version
以下是成功安装后的效果,会直接输出当前的jbrowseCLI的版本号
![]()
3.选择你想要下载Jbrowse项目的路径,并且通过命令下载jbrowse项目
jbrowse create jbrowse2 //下载jbrowse2
接下来你会看到整个项目信息

这就说明你已经成功安装了jbrowse2!
3.启动项目
有两种方式可以启动当前项目:
1.因为整个项目已经是编译过后的项目,可以直接放在nginx的静态资源目录下,直接访问本地nginx配置的地址就可以直接访问到jbrowse基因组浏览器。
2.启动jbrowse2的项目:
npx serve .
# or
npx serve -S . # if you want to refer to symlinked data later on
一定要注意的是,这里启动项目的时候,必须在整个JBrowse2的根目录下启动!
如果进入了项目内部,启动不会成功!
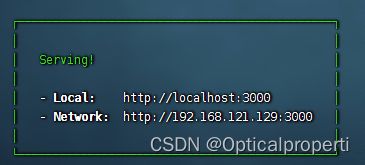
当看到这个界面的时候,jbrowse2就成功启动,此时可以直接通过当前地址进行访问。
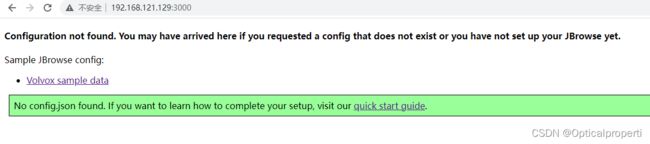
当成功访问到jbrowse2本地项目的时候,应该出来的是这个界面,说明你已经成功启动了该项目!
并且你可以点击 “Volvox sample data” 查看官方给的demo项目。

4.配置本地基因组信息
因为我的本地项目主要是以 .bed .vcf .gff 三个后缀的文件组成的,所以这里只介绍这三种文件的配置。
1.首先通过命令在jbrowse项目中创建自己的文件夹,当前文件夹名根据自己的需求创建
sudo mkdir mydata //当前mydata是文件夹名称。
首先创建文件夹,这里我暂时使用文件夹名为 “mydata” 代替
2.创建 jbrowse 配置的第一步是加载基因组程序集:
如果当前的服务器上没有samtools工具,请在服务器通过该命令下载该工具:
sudo apt install samtools
接下来需要通过我们自己的 FASTA文件生成一个“config.json”文件。
//假设当前fa文件名为 genome.fa
samtools faidx genome.fa
//--out 后面的路径为需要生成config.json文件路径,当前暂时表示为刚才创建的文件夹路径
// 注意!当前的genome.fa文件在 --load copy之后会复制一份到当前指定的文件路径下
jbrowse add-assembly genome.fa --load copy --out /usr/local/jbrowse/mydata/
生成config.json文件成功之后可以看到我们的config.json文件,此时可以重新启动 jbrowse项目查看我们刚刚生成的配置,通过路径地址加上 config.json文件指定路径获取。后文会继续说明如何访问我们自己的配置。
3.添加基因组信息
注意 track配置只能配置一个assembly,如果配置了多个assembly需要指定当前的 assemblyName,那么需要在当前的添加track的代码中加上--assemblyNames hg19(其中hg19是需要指定的assemblyName)
如果当前的服务器上没有samtools工具,请在服务器通过该命令下载该工具:
sudo apt install tabix
VCF格式:
bgzip file.vcf
tabix file.vcf.gz
jbrowse add-track file.vcf.gz --load copy --out /usr/local/jbrowse/mydata/
BED格式:
bgzip file.bed
tabix file.bed.gz
jbrowse add-track file.bed.gz --out /usr/local/jbrowse/mydata/ --load copy
更多格式请参考官方文档:https://jbrowse.org/jb2/docs/quickstart_web/
4.添加成功后重新启动jbrowse2项目,通过路径访问可以直接看到我们的项目
http://ip地址:3000/?config=mydata/config.json
但是可能访问当前的地址并没有对我们的基因组信息直接展示,需要通过点击以下
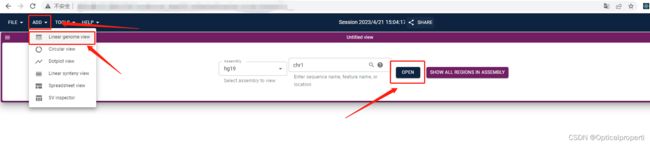

就可以看到我们自己的基因组文件展示出来了。
4.删除配置
在服务器上如果需要删除指定的 track配置的话,需要将当前路径切换到config.json所在的路径,执行以下代码
//其中trackName是config.json中的trackId
jbrowse remove-track trackId

5.配置默认展示
配置默认展示的官方文档地址:https://jbrowse.org/jb2/docs/urlparams/
前面也说过了,访问地址的前半部分是:
http://IP地址:3000/
那么我们可以继续拼接参数以达到我们默认打开jbrowse的页面信息。
配置文件路径: JBrowse 2 配置文件的路径,相对于磁盘上的当前文件夹。我们的例子路径就是在项目中的 mydata文件夹下。
?config=mydata/config.json
指定展示的基因组程序集: 是我们前面配置的assmbly,需要指定在config.json中,assemblyName程序集“名称”,假设当前我配置的其中一个assemblyNames信息中有一个是hg19,则以下面的方式拼接,这仅用于启动单个线性基因组视图。
&assembly=hg19
加载时执行到该区域的导航:
&loc=chr1:6000-7000
//可以有以下几种方式:
chr1:6000-7000 // using - notation for range
chr1:6000..7000 // using .. notation for range
chr1:7000 // centered on this position
加载的基因组文件 :需要指定展示出来的基因组文件,其中名称为 config.json 中自行配置的 track 中的 trackId
&tracks=gene_track,vcf_track
最后我们可以通过将整个访问地址拼接起来,从而访问到 gene_track 在chr1:6000-7000的基因信息了。
http://IP地址:3000/?config=mydata/config.json&assembly=hg19&loc=chr1:6000-7000&tracks=gene_track
接下来你就可以通过路径访问到本地的jbrowse浏览器了,并且可以在开发过程中,通过不同的路径访问达到类似搜索的结果,并通过iframe的方式嵌入自己的浏览器进行展示。