【生物信息学】scRNA-seq数据分析(一):质控~细胞筛选~高表达基因筛选
文章目录
- 一、实验介绍
- 二、实验环境
-
- 1. 配置虚拟环境
- 2. 库版本介绍
- 三、实验内容
-
- 0. 导入必要的库
- 1. 质控
- 2. 细胞筛选
- 3. 高表达基因筛选
一、实验介绍
质控~ 细胞筛选 ~高表达基因筛选
二、实验环境
1. 配置虚拟环境
可使用如下指令:
conda create -n bio python==3.9
conda activate bio
pip install -r requirements.txt
其中,requirements.txt:
numpy==1.21.5
pandas==1.4.4
scanpy==1.9.6
2. 库版本介绍
| 软件包 | 本实验版本 |
|---|---|
| numpy | 1.21.5 |
| pandas | 1.4.4 |
| python | 3.8.16 |
| scanpy | 1.9.6 |
| scipy | 1.10.1 |
| seaborn | 0.12.2 |
三、实验内容
0. 导入必要的库
import numpy as np
import pandas as pd
import scanpy as sc
- Scanpy是一个用于单细胞RNA测序数据分析的Python库,提供了许多功能和工具来处理和分析单细胞数据
1. 质控
# 设置Scanpy参数
sc.settings.verbosity = 3
sc.logging.print_header()
sc.settings.set_figure_params(dpi=80, facecolor='white')
# 定义结果文件路径
results_file = 'write/pbmc3k.h5ad'
# 读取单细胞数据
adata = sc.read_10x_mtx(
'data/filtered_gene_bc_matrices/hg19/', # 数据目录
var_names='gene_symbols', # 使用基因符号作为变量名
cache=True) # 写入缓存文件以便后续更快读取
# 确保基因名唯一
adata.var_names_make_unique()
# 绘制展示高度表达的基因
sc.pl.highest_expr_genes(adata, n_top=20)

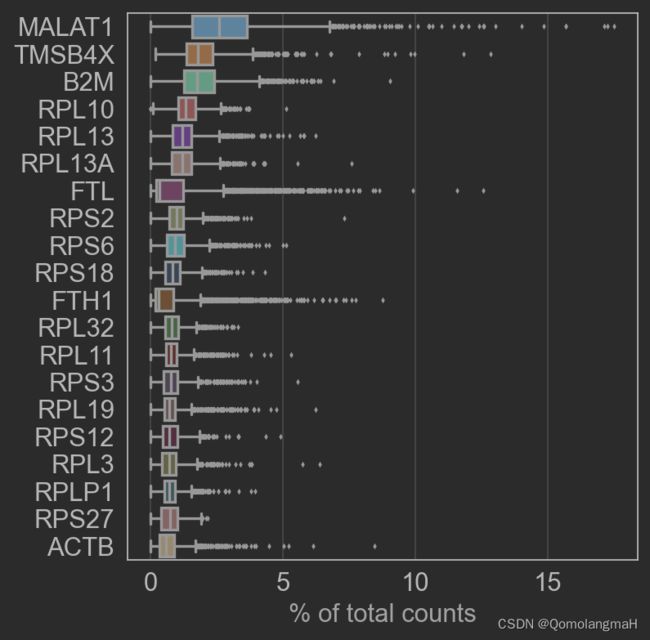
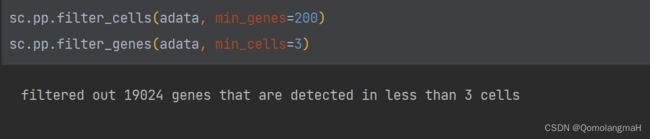
2. 细胞筛选
# 过滤细胞和基因
sc.pp.filter_cells(adata, min_genes=200)
sc.pp.filter_genes(adata, min_cells=3)
# 标记线粒体基因
adata.var['mt'] = adata.var_names.str.startswith('MT-')
# 计算质量控制指标
sc.pp.calculate_qc_metrics(adata, qc_vars=['mt'], percent_top=None, log1p=False, inplace=True)
# 绘制质量控制指标的小提琴图和散点图
sc.pl.violin(adata, ['n_genes_by_counts', 'total_counts', 'pct_counts_mt'], jitter=0.4, multi_panel=True)
sc.pl.scatter(adata, x='total_counts', y='pct_counts_mt')
sc.pl.scatter(adata, x='total_counts', y='n_genes_by_counts')



3. 高表达基因筛选
# 进一步的过滤和归一化
adata = adata[adata.obs.n_genes_by_counts < 2500, :]
adata = adata[adata.obs.pct_counts_mt < 5, :]
# 总计数归一化
sc.pp.normalize_total(adata, target_sum=1e4)
# 对数变换
sc.pp.log1p(adata)
# 特征选择:识别高度变异的基因
sc.pp.highly_variable_genes(adata, min_mean=0.0125, max_mean=3, min_disp=0.5)
# 绘制高度变异基因的图
sc.pl.highly_variable_genes(adata)
# 设置.raw属性
adata.raw = adata
# 实际过滤数据
adata = adata[:, adata.var.highly_variable]
# 数据回归处理和标准化
sc.pp.regress_out(adata, ['total_counts', 'pct_counts_mt'])
sc.pp.scale(adata, max_value=10)
