scenic:单细胞调控网络推理和聚类
这是GRN分析中scenic的文献,发表在2017年的nature methods(SCENIC : single-cell regulatory network inference and clustering),学习了解其原理。
摘要
我们提出了scenery,一种用于从单细胞 rna-seq 数据中同时进行基因调控网络重建和细胞状态识别的计算方法 (http://scenic.aertslab.org)。根据肿瘤和大脑的单细胞数据概要,我们证明顺式调控分析可用于指导转录因子和细胞状态的识别。scenic为驱动细胞异质性的机制提供了重要的生物学见解。
介绍
细胞的转录状态产生于潜在的基因调控网络(GRN),其中有限数量的转录因子(TF)和辅因子相互调节及其下游靶基因。单细胞转录组分析的最新进展为高分辨率识别转录状态和状态之间的转换(例如分化期间)提供了令人兴奋的机会。针对单细胞 RNA-seq 优化的统计技术和生物信息学方法带来了新的生物学见解,但目前尚不清楚是否可以确定稳定细胞状态下特定且稳健的 GRN。这确实可能具有挑战性,因为在单细胞水平上,由于转录爆发和其他来源的基因表达的随机变化,基因表达可能与 TF 输入的动态部分脱节。已经开发了一些从单细胞 RNAseq 数据推断共表达网络的方法,但这些方法不使用调控序列分析来预测 TF 和靶基因之间的相互作用。
我们推断将顺式调控序列与单细胞基因表达连接可以克服丢失和技术变异,从而优化细胞状态的发现和表征。为此,我们开发了单细胞调控网络推理和聚类(SCENIC)来绘制GRN,然后通过评估每个细胞中GRN的活性来识别稳定的细胞状态。 SCENIC 工作流程由三个步骤组成(图 1a,Supplementary Fig. 1 and see Online Methods)。

(a) In the SCENIC workflow, coexpression modules between TFs and candidate target genes are first inferred using GENIE3 or GRNBoost. RcisTarget then identifies modules for which the regulator’s binding motif is significantly enriched across the target genes and creates regulons with only direct targets. AUCell scores the activity of each regulon in each cell, thereby yielding a binarized activity matrix. The prediction of cell states is based on the shared activity of regulatory subnetworks.
(a) 在 SCENIC 工作流程中,首先使用 GENIE3 或 GRNBoost 推断 TF 和候选目标基因之间的共表达模块。然后,RcisTarget 识别调节器的结合基序在目标基因上显着富集的模块,并创建仅具有直接目标的调节子。 AUCell 对每个细胞中每个调节子的活性进行评分,从而产生二值化的活性矩阵。细胞状态的预测基于调节子网络的共享活动。

(a) 第一步,使用 GENIE3(随机森林)或 GRNBoost(梯度增强)推断转录因子和候选靶基因之间的共表达模块。每个模块由一个转录因子及其预测目标组成,纯粹基于共表达。 (b) 第二步,用 RcisTarget 分析每个共表达模块,以识别富集的基序;仅保留 TF 基序富集的模块和靶标。每个 TF 及其潜在的直接目标都是一个调节子。 © 在第三步中,使用 AUCell 评估每个细胞中每个调节子的活性,AUCell 计算恢复曲线下的面积,整合调节子中所有基因的表达等级。 AUCell 分数用于生成调节子活动矩阵。该矩阵可以通过为每个调节子设置 AUC 阈值来二值化,这将确定调节子在哪些细胞中“开启”。 (d) 调节子活动矩阵可用于对细胞进行聚类(例如 t-SNE),从而根据调节子网络的共享活动来识别细胞类型和状态。
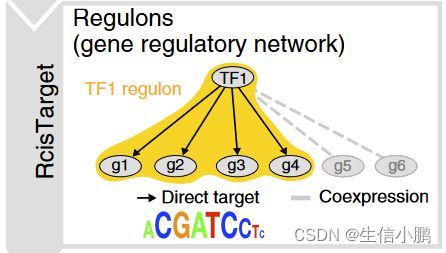
第一步,使用 GENIE3 识别与 TF 共表达的基因组(补充图 1a)。由于 GENIE3 模块仅基于共表达,因此它们可能包含许多误报和间接目标。为了识别假定的直接结合靶标,每个共表达模块均使用 RcisTarget 进行顺式调控基序分析(补充图 1b 并参见在线方法)。仅保留具有显着富集正确上游调节因子的基序的模块,并且对它们进行修剪以去除缺乏基序支持的间接目标。我们将这些经过处理的模块称为调节子。
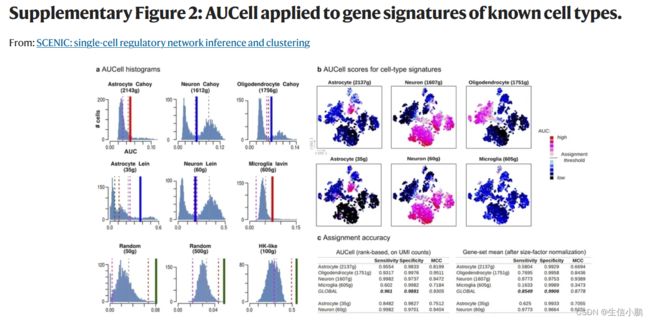
作为 SCENIC 的一部分,我们开发了 AUCell 算法来对每个细胞中每个调节子的活性进行评分(补充图 1c 和 2)。**对于给定的调节子(regulon),比较细胞间的 AUCell 分数可以识别哪些细胞具有明显更高的子网络活动。**生成的二元活动矩阵的维数降低了,这对于下游分析非常有用。例如,基于该矩阵的聚类根据监管子网的共享活动来识别细胞类型和状态。由于调节子是作为一个整体进行评分的,而不是使用单个基因的表达,因此这种方法对于 dropout 具有鲁棒性(补充图 3)。
为了评估 SCENIC 的性能(performance),我们将其应用于包含来自成年小鼠大脑的已知细胞类型的 scRNA-seq 数据集(图 1b-e)。该分析提供了 1,046 个初始共表达模块中的 151 个调节子,其中相应 TF 的基序显着富集(初始 TF 的 7%)。对每个细胞的调节子活性进行评分揭示了预期的细胞类型(图1d,e)以及每种细胞类型的潜在主调节子列表(例如补充图4中的小胶质细胞网络)。按细胞类型进行聚类(总体灵敏度为 0.88,特异性为 0.99,调整兰德指数 (ARI) > 0.80)比许多专用的单细胞聚类方法更准确。
为了评估 SCENIC 的稳健性(robustness),我们重新分析了小鼠大脑数据:完整的数据集; 100 个随机选择的细胞样本来模拟小数据集;或三分之一的测序读数来模拟低覆盖率数据。 SCENIC 识别出仅由少数细胞代表的细胞类型(例如,来自小胶质细胞、星形胶质细胞或中间神经元的两到六个细胞;补充图 5)。此外,预测的 TF 与细胞类型的关联与之前建立的一致角色(图 1c),并且这种准确性优于标准分析管道(补充图 3e)。

还应用 SCENIC 来识别少突胶质细胞瘤 (来自 6 个肿瘤的 4,043 个细胞)和黑色素瘤 (来自 14 个病变的 1,252 个细胞)的 scRNA-seq 数据集中的复杂细胞状态。由于肿瘤特异性突变和复杂的基因组畸变,癌细胞状态的识别比正常细胞状态更具挑战性15。标准聚类根据肿瘤来源对细胞进行分组(图 3a、b),但 SCENIC 揭示了不同的情况。对于少突胶质细胞瘤,肿瘤中存在三种癌细胞状态(图 3c-e),每种状态均由预期的 TF 驱动,包括少突胶质细胞样状态的 SOX10/4/8、OLIG1/2 和 ASCL1; SOX9、NFIB 和 AP-1 用于星形胶质细胞样状态; E2F 和 FOXM1 用于循环细胞。此外,将扩散图应用于二元 SCENIC 矩阵(补充图 8)重建了从干细胞样分支到少突胶质细胞样和星形胶质细胞样分支的分化轨迹。请注意,与正常少突胶质细胞分化相比,这条路径代表了不同的“轨迹”(参见补充图 9 对 5,069 个少突胶质细胞的 SCENIC 分析)。我们在黑色素瘤数据上观察到类似的肿瘤效应校正,其中 SCENIC 识别了肿瘤中的细胞组(补充图 10),包括由与少突胶质细胞瘤中类似的 TF 驱动的循环细胞簇(例如,E2F1/2/8 和MYBL2;图 3f-h 和补充图 10)。与 Combat16 和 Limma17 等专门的批量效应消除方法相比,这些方法需要先验地指定批量效应的来源(补充图 11),SCENIC 通过使用生物驱动的特征自动消除肿瘤效应。
补充部分不细看了,重点看使用方法和算法的原理
online methods
SCENIC workflow
SCENIC 是一个基于三个新 R/bioconductor 包的工作流程:(i) GENIE3,基于共表达识别潜在的 TF 目标; (ii) RcisTarget,执行 TFmotif 富集分析并确定直接目标(调节子 regulons); (iii) AUCell,对单细胞上的调节子(或其他基因组)的活性进行评分。我们还提供在 Spark上实现的 GRNBoost,作为在更大数据集上构建共表达网络的可扩展替代方案(步骤 i,替换 GENIE3)。三个 R/bioconductor 包和 GRNBoost 包括详细的教程,以方便它们在自动化 SCENIC 管道中的使用,以及独立的工具。
GENIE3
GENIE3。 GENIE3是一种从基因表达数据推断基因调控网络的方法。简而言之,它训练随机森林模型来预测数据集中每个基因的表达,并使用 TF 的表达作为输入。然后使用不同的模型得出 TF 的权重,测量它们各自与预测每个目标基因表达的相关性。最高权重可以转化为 TF 目标监管链接。由于 GENIE3 使用随机森林回归,因此它具有允许 TF 与其候选目标之间存在复杂(例如非线性)共表达关系的附加值。 GENIE3 可在 Python、Matlab 和 R 中使用。为了包含在 SCENIC 工作流程中,我们优化了 GENIE3 之前的 R 实现。这个新实现的核心现在是用 C 编写的(这使其速度快了几个数量级),它需要更少的内存,并且支持并行执行。 GENIE3 是 DREAM4 和 DREAM5 挑战赛中表现最好的网络推理方法24。新包在 DREAM 挑战中提供了与之前现有实现类似的结果,但速度有所提高。The comparison is available at the following website: http://www. montefiore.ulg.ac.be/~huynh-thu/GENIE3.html.
GENIE3 的输入是一个表达矩阵。首选表达值是基因汇总计数(可能使用也可能不使用唯一分子标识符 UMIs)。其他测量值,例如每百万计数或转录本 (TPM) 和 FPKM/RPKM 也可以作为输入接受。然而,请注意,第一个网络推理步骤基于共表达,一些作者建议避免此任务的样本内标准化(即 TPM),因为它们可能会引起人为共变。为了评估输入矩阵的归一化在多大程度上影响 SCENIC 的输出,我们还在库大小归一化后在 Zeisel 等人数据集上运行了 SCENIC(使用 scran的标准管道,该管道执行簇内大小-因子归一化)。结果具有高度可比性,无论是在生成的簇或细胞类型方面(从原始 UMI 计数或归一化计数获得的细胞类型之间的 ARI:0.90,与作者的细胞类型相比,从归一化计数获得的 ARI:0.87)以及识别的 TF组(图 1b 中突出显示的 30 个调节子中的 26 个)。此外,在该项目过程中,我们将 GENIE3 应用于多个数据集,其中一些具有 UMI 计数(例如,小鼠大脑和少突胶质细胞),其他具有 TPM(例如,人脑和黑色素瘤),并且两个单元都提供了可靠的结果。
GENIE3 的输出是一个表格,其中包含基因、潜在调节因子及其**“重要性度量”(IM),它代表TF(输入基因)在目标预测中的权重**。我们探索了几种确定阈值的方法(例如,查看使用 RcisTarget 修剪后的排名、分布和输出),最终选择为每个 TF 构建潜在目标的多个基因集:(i)设置多个 IM 阈值(IM > 0.001) IM > 0.005),(ii) 为每个 TF 选取 50 个具有最高 IM 的目标,以及 (iii) 为每个目标基因仅保留前 5、10 和 50 个 TF(然后按 TF 分割)。在所有这些情况下,仅考虑 IM > 0.001 的链接。此外,每个基因集随后被分为正相关和负相关目标(即 TF 和潜在目标之间的 Spearman 相关性),以区分可能激活的目标和抑制的目标。最后,仅保留至少具有20个基因的基因集(TF共表达模块)用于下一步。
GRNBoost
GRNBoost 基于与 GENIE3 相同的概念:纯粹从基因表达矩阵推断每个目标基因的调节因子。然而,GRNBoost 使用 XGBoost 库中的梯度提升机 (GBM)实现来实现这一点。 GBM 是一种集成学习算法,它使用 boosting作为一种策略,将多个弱学习器(如浅树)组合成强学习器。这与 GENIE3 使用的随机森林方法形成鲜明对比,后者使用装袋(引导聚合)进行模型平均以提高回归精度。 GRNBoost 使用梯度增强树桩(深度为 1 的回归树)作为基础学习器。 GRNBoost 的主要贡献是将这种多重回归方法转换为基于 Apache Spark23 的 Map/Reduce32 框架。在 GRNBoost 中,核心数据条目是基因名称的元组和基因表达值的向量。 GRNBoost 使用 Spark RDD,首先将基因表达向量划分到计算集群中可用的节点上。随后,它构建一个预测矩阵,其中包含所有候选调节基因的表达值。使用 Spark 广播变量,将预测矩阵广播到不同的计算分区。在框架的映射阶段,GRNBoost 迭代基因元组(表达向量),并使用预测矩阵来训练 XGBoost 回归模型,并将表达向量作为各自的训练标签。从经过训练的模型中,提取调节器-目标关系的强度并将其作为一组网络边缘发出。在reduce阶段,所有的边集都被组合到最终的监管网络中。
GRNBoost 和 GENIE3 的性能在配备 2 个 Intel Xeon E2696 V4 CPU 的工作站上进行比较,该 CPU 总共有 44 个物理核心或 88 个线程以及 128 GB 2133Ghz ECC 内存。大数据集和大预测矩阵会导致网络推理受到内存限制而不是 CPU 限制。为了轻松地将所需的内存量放入可用的 128 GB 内存中,我们将分区数量减少到 11 个,因此最多只有 11 个同时运行的预测矩阵。然而,我们将每个单独的 XGBoost 回归可用的线程数量增加到 8 个,从而有效地使用工作站中的所有可用线程 (88)。 GRNBoost 采用 Scala 编程语言编写,可用作软件库或从命令行作为 Spark 作业提交。
这一段涉及到挺多算法知识,我觉得不需要深入纠结
RcisTarget
RcisTarget 是 i-cisTarget 和 iRegulon 基序富集框架的新 R/Bioconductor 实现。RcisTarget 识别基因列表中丰富的 TF 结合基序和候选转录因子。简而言之,RcisTarget 基于两个步骤。首先,它选择在基因集中基因的转录起始位点 (TSS) 周围显着过度表达的 DNA 基序。这是通过在数据库上应用基于恢复的方法来实现的,该数据库包含每个基序的全基因组跨物种排名。注释到相应 TF 并获得归一化富集分数 (NES) > 3.0 的基序将被保留。接下来,对于每个基序和基因集,RcisTarget 预测候选目标基因(即基因集中排名在前沿之上的基因)。该方法基于 Aerts 等人描述的方法,该方法也在 i-cisTarget(Web 界面)和 iRegulon(Cytoscape 插件)35 中实现。因此,当使用相同的参数和数据库时,RcisTarget 提供与 i-cisTarget 或 iRegulon 相同的结果,并与 Janky 等人中的其他 TFBS 富集工具进行基准测试 。文档中给出了有关该方法及其在 R 中的实现的更多详细信息。
为了构建最终的调节子,我们合并了每个 TF 模块的预测目标基因,这些基因显示了给定 TF 的任何基序的富集。为了检测抑制,理论上可以对负相关 TF 模块采用相同的方法。然而,在我们分析的数据集中,这些模块数量较少,并且显示出非常低的主题富集度。因此,我们最终决定从工作流程中排除直接抑制的检测,并仅继续处理正相关目标。本文中用于分析的数据库是来自 iRegulon(基于基因的基序排名)的人类和小鼠的“18k 基序集合”。对于每个物种,我们使用两个基因基序排名(TSS 周围 10 kb 或 TSS 上游 500 bp),这决定了 transcTSS 周围的搜索空间。
AUCell
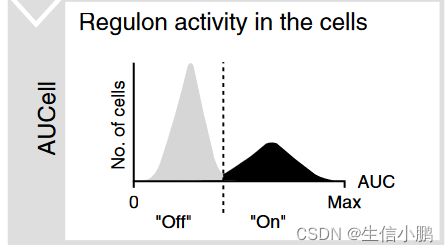
AUCell 是一种新方法,使研究人员能够在单细胞 RNAseq 数据中识别具有活跃基因调控网络的细胞。 AUCell 的输入是基因集,输出是每个细胞中基因集的“活性”(gene set ‘activity’)。在 SCENIC 中,这些基因集是调节子,由 TF 及其假定目标组成。 AUCell 将调节子的富集计算为特定细胞中所有基因排名的恢复曲线下面积 (AUC),从而根据基因的表达值对基因进行排名。因此,该方法独立于基因表达单位和标准化程序。此外,由于细胞是单独评估的,因此可以轻松应用于更大的数据集(例如,如果需要,可以对表达矩阵进行子集化)。简而言之,评分方法基于回收率分析,其中 x 轴(补充图 1c)是基于表达水平的所有基因的排名(具有相同表达值的基因,例如“0”,被随机排序) ); y 轴是从输入集中恢复的基因数量。然后,AUCell 使用 AUC 来计算输入基因集的关键子集是否在每个细胞的排名顶部富集。**通过这种方式,AUC代表了特征中表达基因的比例以及它们与细胞内其他基因相比的相对表达值。**此步骤的输出是一个矩阵,其中包含每个细胞中每个基因集的 AUC 分数。我们直接使用 AUC 分数(跨调节子)作为连续值来对单个细胞进行聚类,或者使用每个调节子的 AUC 分数的截止值生成二进制矩阵。
这些截止值要么自动确定,要么通过检查 AUC 分数的分布来手动调整。补充图 2a 中提供了 AUC 分布的一些示例。补充图 2b、c 显示了使用先前发布的神经元和神经胶质基因特征对 AUCell 进行的验证。包中包含的教程还包括该方法每个步骤的实用解释和含义。
Cell clustering based on gene regulatory networks 基于基因调控网络的细胞聚类
细胞调节子活性总结在矩阵中,其中列代表细胞,行代表调节子。在二元调节子活动矩阵中,对应于给定单元中的活动调节子的矩阵坐标将包含“1”,否则包含“0”。等效矩阵包含每个细胞调节子的连续 AUC 值,通常称为 AUC 活动矩阵。任一调节子活动矩阵的聚类揭示了在细胞子集中经常活动的调节子组(共同形成网络)。二元活动矩阵倾向于突出细胞之间的高阶相似性(因此大大减少批次效应和技术偏差);另一方面,AUC矩阵可以让研究人员观察到更细微的变化。对于可视化,我们主要使用 tSNE(Rtsne package,我们总是测试多个困惑度值和距离度量/PC 数量的一致性)和具有分层聚类的热图(尽管热图具有选定的调节器,但 t-SNE 始终运行整个矩阵)。在教程中,我们还提供了几个探索结果的选项。例如,如何检测最可能的稳定状态(t-SNE 中的较高密度区域),并帮助识别关键调节因子、已知细胞特性(基于数据集注释)和 GO 术语(t-SNE 中基因的 GO 富集分析)调节子簇)可能与检测到的状态相关。
SCENIC runs on the different data sets
可以直接翻阅tutorial
Gene filtering
对于运行 GENIE3 的基因过滤,我们应用了基于基因计数总数和检测到该基因的细胞数量的软过滤器。第一个过滤器是每个基因的读取总数,旨在删除最有可能不可靠且仅提供噪声的基因。具体值取决于数据集;对于本文中使用的阈值,我们将阈值设置为例如 3 个 UMI 计数(略高于非零值的中位数)乘以数据集中细胞数量的 1%(例如,在小鼠大脑中:3 UMI 计数 × 30(细胞的 1%)= 每个基因至少 90 个计数)。第二个过滤器,即检测到基因的细胞数量(例如,>0 UMI 或 >1 log2(TPM)),是去除仅在一个或极少数细胞中表达的基因(它们会获得很多如果它们碰巧在给定的单元格中重合,则为权重)。在工作流程中,我们建议将第二次过滤设置为低于要检测的最小细胞群。例如,由于小胶质细胞约占数据集中总细胞的 3%,因此我们使用至少 1% 的细胞的检测阈值。
总结一下
这里不是原文内容,摘自其他文章
原文涉及内容多,主要内容再总结一下
SCENIC分析原理
在输入单细胞基因表达量矩阵后,SCENIC经过以下三个步骤完成转录因子分析:第一步是构建共表达网络、第二步是构建TF-targets网络、第三步是计算Regulons活性,每一个步骤都由一个专门的软件包完成。
第一步:GENIE3——共表达网络构建
第一步由GENIE3或GRNBoost软件完成,这里以GENIE3为例介绍。GENIE3 (GEne NetworkInference with Ensemble of trees) ,基于树的基因网络推理,是一种从基因表达数据推断基因调控网络的方法。软件以单细胞基因表达量矩阵为输入文件,以每个目标基因 (gene) 为输出,以转录因子 (TF) 为输入,构建P个随机森林树(P=矩阵中基因数量),并计算每个TF与gene之间的重要性评分 (IM) ,最终可以获得TF-genes共表达模块。最后删除IM低于阈值的基因关系,过滤基因数低于50的模块。

第二步:RcisTarget——motif富集及靶基因预测
从第一步获得了TF-genes共表达网络,但这个网络只是基于TF和gene表达量相关性推测的,TF和gene之间是否现实存在调控关系还需要进一步确证。确证的方法主要从TF功能结构入手,从图3可以看出,TF是通过直接与DNA特定位置序列而发挥作用的,因此可以通过反向查看gene上是否存在TF结合的motif序列来验证TF与gene的靶向关系。
这一步可以借助RcisTarget软件完成,该软件运行必备两个数据库:1)gene-motif排名数据库:为每个motif提供所有gene的排名(~分数);2)motif-TF注释数据库:对每一个motif注释其所对应的TF。由于不同物种基因组不一样,导致每个motif对应靶基因不同,因此针对不同物种需要构建不同的数据库,软件目前配置了人、小鼠、果蝇数据库.
那么具体验证过程,首先基于gene-motif数据库,每个motif对模块中所有基因进行累积,模块中的基因排名越靠前,累积曲线越高,曲线下面积 (AUC) 越大,表明motif在该模块中的富集程度越高,然后对每个模块选取显著富集的motif,并预测其靶基因,最终综合TF-genes模块和靶基因预测结果,构成一个包含了TF和靶基因的基因调控网络模块 (regulons)。
第三步:AUCell——Regulons活性定量
第三步就是Regulons活性定量。这一步由AUCell软件完成,AUCell是一种新的方法,允许在scRNA-seq数据中识别具有活性基因调控网络的细胞。
实际分析过程中,输入到AUCell的是一个基因集,输出的是每个细胞中的基因集“活性” (AUC, Area Under Curve)。在SCENIC中,这些基因集即Regulons中所有基因,针对每个细胞,将细胞中所有基因按照表达量从高到低进行排序,根据Regulons中的基因在序列中的位置,计算累计曲线面积 (AUC) ,即为Regulons在细胞中的活性。
学习文献
Aibar, S., González-Blas, C., Moerman, T. et al. SCENIC: single-cell regulatory network inference and clustering. Nat Methods 14, 1083–1086 (2017). https://doi.org/10.1038/nmeth.4463

单细胞个性化分析之转录因子篇