生信工作流框架搭建 | 02-nextflow 实战
目录
- 生信工作流框架搭建 | 02-nextflow
-
- 前情提要
- 开始使用
-
- 依赖
- 安装
- 核心概念
-
- 一个fastqc的示例,加深理解
- 快速搭建你的程序
-
- 你需要仔细阅读的:
- 可以快速浏览(但需要知道大概有什么,以便后来查览):
- 报错!你会踩哪些坑?
- 下期预告
生信工作流框架搭建 | 02-nextflow
本篇为biodoge《生信工作流框架搭建》系列笔记的第3篇,该系列将持续更新。
前情提要
上回生信工作流框架搭建 | 01-nextflow、snakemake、wdl 对比测试为大家对比了三种主流工作流语言,根据实际情况,我选择了nextflow为后续搭建流程。那么,本篇即为nextflow的实用教程。
注意,nextflow官方教程已经非常详细,我不会一点点把教程搬过来,只从实战角度快速上手。
本文示例基于AWS EC2环境。
开始使用
ok,让我们一起打开 https://www.nextflow.io/ 开始使用吧!
依赖
- 任何POSIX兼容系统(Linux,OS X等)
- Bash 3.2(或更高版本)
- Java 11(或更高版本,最多 18)。
安装
#安装java(版本11-18)首先要看是否已经安装过了java环境
$ java -version
#到java官网找合适版本 wget,我这里找的是java17(不同版本有差别,可以查一查)
$ mkdir java
$ cd java
$ wget https://download.oracle.com/java/17/latest/jdk-17_linux-x64_bin.tar.gz
$ tar -zxvf jdk-17_linux-x64_bin.tar.gz
#配环境变量
$ vi /etc/profile
#输入以下内容
insert:#set java environment
export JAVA_HOME=/usr/java/jdk1.7.0_79
export CLASSPATH=.:$JAVA_HOME/lib/dt.jar:$JAVA_HOME/lib/tools.jar
export PATH=$PATH:$JAVA_HOME/bin
export JAVA_HOME CLASSPATH PATH
#wq
$ source /etc/profile
#使用conda安装nextflow
$ conda install nextflow
核心概念
在详细阅读官方文档前,我们要了解一点:
工作流语言/框架是在管道的功能逻辑和底层执行系统之间提供了抽象。
那么这层抽象是怎样的结构呢?
通道channel、流程process、工作流workflow

一个fastqc的示例,加深理解
无论是RNAseq还是宏基因组分析,首先都会经过质控这个步骤,且往往都是数据处理的第一步。
如果我们想搭建一个fastqc的小工作流,该如何实现呢?
我的思路是,无论何种工作流语言,核心都是输入输出。
第一步,确定输入:sample_R1.fastq.gz sample_R2.fastq.gz,我们也可以批量处理样本:sample1、sample2、sample3……
确定输出:我们需要的fastq运行结果:sample.zip sample.html
第二步,确定传输方式。HOW——我们如何读取fastq文件?——nextflow贴心准备了fromFilePairs方法(颗粒度真的很细!),在fromFilePairs里面传入一个参数(可以在顶部定义,这样想更改文件路径的时候直接修改顶部参数即可,非常棒的是可以识别通配符),通过该方法,我们会读入一个tuple,形如
| sampleID | reads |
|---|---|
| sample1 | [/some/data/sample1_R1.fastq.gz , /some/data/sample1_R2.fastq.gz] |
| sample2 | [/some/data/sample2_R1.fastq.gz , /some/data/sample2_R2.fastq.gz] |
传入process时,我们要用tuple val(xx),path(yy)这样的形式,xx和yy在脚本中正确调用就好。
我们如何获取输出文件?——publishDir指定即可。
第三步,把process的骨架搭起来——输入输出脚本,加上一些指令——最后工作流调用任务
// Declare syntax version 版本号很关键,不同版本语法不同!推荐大家使用dsl2
nextflow.enable.dsl=2
// Script parameters 脚本参数在这里定义
params.read = "/some/data/*.fastq.gz" //可以识别通配符
process fastq {
container "${ workflow.containerEngine == 'singularity' ?
'https://depot.galaxyproject.org/singularity/fastqc:0.11.9--0' :
'quay.io/biocontainers/fastqc:0.11.9--0'}" //指定容器
publishDir "/result/",mode:'copy'
input: //输入 这里是tuple
tuple val(meta),path(reads)
output: //输出 也许工作目录会有很多结果文件,但我们只需要特定的输出文件,那么只有这个指定的文件会被视为需要输出的
path("${id}/*.html"),emit:html //提问,这里的emit有什么用?
path("${id}/*.zip"),emit:zip
when:
task.ext.when == null || task.ext.when
script: //脚本,linux命令,与命令行执行基本一样,除了要注意一些字符需要反转义,系统变量前也要加/反转义
"""
mkdir ${id}
fastqc -o ${id} -t 16 ${reads}
"""
}
workflow {
reads = Channel.fromFilePairs(params.read) //通过通道传递参数,读取文件,fromPair这个方法可以直接读成对reads
FASTQC(reads) //像调用函数那样调用任务,括号传参
}
当然我们处理更复杂的数据时,我们完全可以采用fromPath的方法导入一个csv文件(此时必须要split.csv()操作符协助),如下,这样可以分别调用reads1、2
| sampleID | reads1 | reads2 |
|---|---|---|
| sample1 | /some/data/sample1_R1.fastq.gz | /some/data/sample1_R2.fastq.gz |
| sample2 | /some/data/sample2_R1.fastq.gz | /some/data/sample2_R2.fastq.gz |
nextflow.enable.dsl=2
params.read = "/some/data/*.fastq.gz"
process fastq {
container "${ workflow.containerEngine == 'singularity' ?
'https://depot.galaxyproject.org/singularity/fastqc:0.11.9--0' :
'quay.io/biocontainers/fastqc:0.11.9--0'}" //指定容器
publishDir "/result/",mode:'copy'
input:
tuple val(id),path(reads1),path(reads2)
output:
path("${id}/*.html"),emit:html
path("${id}/*.zip"),emit:zip
when:
task.ext.when == null || task.ext.when
script: //脚本,linux命令,与命令行执行基本一样,除了要注意一些字符需要反转义,系统变量前也要加/反转义
"""
mkdir ${id}
fastqc -o ${id} -t 16 ${reads1} ${reads2}
"""
}
workflow {
data = channel.fromPath(params.reads)
.splitCsv(header:true)
.view() //这里加个view可以输出data的形式
FASTQC(data)
}
快速搭建你的程序
看一看官方文档,哇,内容好多!我们都要记下来吗?
一个坑的地方:虽然更新了DSL2但前面的很多示例没有同步!而DSL2与原生语法差距又很大,大家要注意!

实则不然,有些内容粗略带过,并不影响我们的使用。特别是刚开始入门,应该先搭建起来,而不是在各种晦涩概念中眼花缭乱,眉毛胡子一把抓。
你需要仔细阅读的:
Get started
Basic concepts
Processes
Channels
Operators ● 它推荐的那几个操作符 (本页是所有 Nextflow 运算符的综合参考。但是,如果您是新手,对于 Nextflow,以下是一些建议的运算符,用于常见用例)
Configuration
DSL2
FAQ 这货不在侧边栏索引,是隐藏条目,但很有用……
Tracing & visualisation 只需要nextflow run命令加上-with-report / -with-timeline / -with-dag,就会有神奇的事情发生
可以快速浏览(但需要知道大概有什么,以便后来查览):
Nextflow scripting 像是Groovy语法的简洁教程……闭包这个东西现在仍然没搞懂
Executors 后续要仔细看你要使用的执行器,比如AWS,本地跑可以忽略……
Operators 其他部分
这里可以看一些博客,毕竟是翻译过来的中文理解起来会方便一些,但是会存在时效性的问题*(官方文档在不断更新)
安利一个nextflow专栏,操作符之类的讲得很清楚。Nextflow编码实践这篇列举了很多编码技巧。
报错!你会踩哪些坑?
- 语法问题
process assembly {
‘{’
^
语法问题。看起来跟括号有关,实际上只能定位到该process,可能是process内某一个指令或脚本写错了语法
- 配置文件无法识别动态指令
Unknown method invocation
multiplyon Integer type
未知方法在整型上调用‘×’
一般在运行process之前发现,可能是config文件中出现了不应该的×(config无法识别动态指令)
- Script compilation error
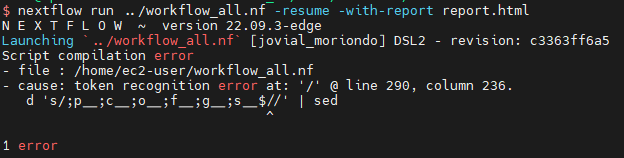
单引号里的任何字符都会原样输出,单引号字符串中的变量是无效的;单引号字串中不能出现单引号(对单引号使用转义符后也不行)
双引号里可以有变量,允许变量替换;双引号里可以出现转义字符
引号内$会被识别为nextflow的变量,而系统变量则需要转义\,同理单纯的/也需要转义\
script:
"""
echo $params.input ->(nextflow参数)
echo \$PWD ->(系统变量)
"""
下期预告
将带大家实践AWS batch+nextflow。如果喜欢的话请大力催更哦