深度学习基础(三)——优化算法
- 1 优化算法
- 1.1 局部最小值点
- 1.2 鞍点
- 2 梯度下降法和随机梯度下降法
- 2.1 梯度下降法
- 2.1.1 一维梯度下降法
- 2.1.2 学习率
- 2.1.3 多维度梯度下降
- 2.2 随机梯度下降法
- 2.3 小批量随机梯度下降法
- 2.1 梯度下降法
- 3 动量法
- 3.1 指数加权平均(EMA)
- 3.2 由指数加权移动平均理解动量法
- 3.2 Nesterov
- 4 Adagrad
- 5 RMSprop
- 6 Adadelta
- 7 Adam
- 8 牛顿法
- 9 拟牛顿法
- 参考
1 优化算法
1.1 局部最小值点
对于目标函数 f(x),如果 f(x) 在 x 上的值比在 x 邻近的其他点的值更小,那么 f(x) 可能是一个局部最小值(local minimum)。如果 f(x) 在 x 上的值是目标函数在整个定义域上的最小值,那么 f(x) 是全局最小值(global minimum)。
深度学习模型的目标函数可能有若干局部最优值。当一个优化问题的数值解在局部最优解附近时,由于目标函数有关解的梯度接近或变成零,最终迭代求得的数值解可能只令目标函数局部最小化而非全局最小化。
虽然找到目标函数的全局最优解很难,但这并非必要。
1.2 鞍点
黑塞矩阵(Hessian Matrix),又译作海森矩阵、海瑟矩阵、海塞矩阵等,是一个多元函数的二阶偏导数构成的方阵,描述了函数的局部曲率。

- 当函数的黑塞矩阵在梯度为零的位置上的特征值全为正时,该函数得到局部最小值。
- 当函数的黑塞矩阵在梯度为零的位置上的特征值全为负时,该函数得到局部最大值。
- 当函数的黑塞矩阵在梯度为零的位置上的特征值有正有负时,该函数得到鞍点。
鞍点更常见:随机矩阵理论告诉我们,对于一个大的高斯随机矩阵来说,任一特征值是正或者是负的概率都是 0.5。那么,以上第一种情况的概率0.5k。由于深度学习模型参数通常都是高维的(k 很大),目标函数的鞍点通常比局部最小值更常见。
2 梯度下降法和随机梯度下降法
2.1 梯度下降法
2.1.1 一维梯度下降法
- 函数值确保降低
- 直到导数为0
假设连续可导的函数 f:R→R 的输入和输出都是标量。给定绝对值足够小的数 ϵ,根据泰勒展开公式:
f(x+ϵ)≈f(x)+ϵf′(x)
接下来我们找一个常数η>0,使得 |ηf′(x)|足够小,那么可以将ϵ替换为 −ηf′(x)得到:
f(x−ηf′(x))≈f(x)−ηf′(x)2.
f(x−ηf′(x))≲f(x).
这意味着,如果我们通过以下规则来更新x:x←x−ηf′(x),函数 f(x) 的值可能被降低。一般来说,我们选取一个初始值x和常数 η>0,然后不断的通过上式来迭代 x,直到达到停止条件,例如 f′(x)2 的值已经足够小。
2.1.2 学习率
上述梯度下降算法中的正数η通常叫做学习率。这是一个超参数,需要人工设定。如果使用过小的学习率,会导致 x 更新缓慢从而需要更多的迭代才能得到较好的解。
2.1.3 多维度梯度下降
假设目标函数 f:Rd→R 的输入是一个 d 维向量x=[x1,x2,…,xd]⊤。目标函数 f(x) 有关 x 的梯度是一个由 d个偏导数组成的向量:
∇xf(x)=[∂f(x)∂x1,∂f(x)∂x2,…,∂f(x)∂xd]⊤.
∇f(x)代替∇xf(x)。梯度中每个偏导数元素∂f(x)/∂xi 代表着 f在 x 有关输入 xi的变化率。为了测量 f 沿着单位向量 u(即 ∥u∥=1)方向上的变化率,在多元微积分中,我们定义f在 x 上沿着 u 方向的方向导数为:
Duf(x)=limh→0f(x+hu)−f(x)h.
该方向导数可以改写为:
Duf(x)=∇f(x)⋅u.
方向导数 Duf(x) 给出了f 在 x 上沿着所有可能方向的变化率。为了最小化f,我们希望找到f 能被降低最快的方向。因此,我们可以通过单位向量 u来最小化方向导数 Duf(x)。
由于 Duf(x)=∥∇f(x)∥⋅∥u∥⋅cos(θ)=∥∇f(x)∥⋅cos(θ), 其中 θ为梯度∇f(x) 和单位向量 u 之间的夹角,当 θ=π,cos(θ) 取得最小值 −1。因此,当 u 在梯度方向∇f(x) 的相反方向时,方向导数 Duf(x) 被最小化。所以,我们可能通过下面的梯度下降算法来不断降低目标函数 f 的值:
x←x−η∇f(x).
2.2 随机梯度下降法
diss:通常目标函数是训练样本上损失函数的平均
f(x)=1nn∑i=1fi(x).
如果使用梯度下降,每次自变量迭代的计算开销为 O(n),它随着 n线性增长。因此,当训练数据样本数很大时,梯度下降每次迭代的计算开销很高。
随机梯度下降(stochastic gradient descent,简称 SGD)减少了每次迭代的计算开销。
在随机梯度下降中,每次迭代我们随机均匀采样一个样本索引 i∈[1,n],并计算梯度 ∇f_i(x) 来迭代 x:
\boldsymbol{x} \leftarrow \boldsymbol{x} - \eta \nabla f_i(\boldsymbol{x}).
这里 η 同样是学习率。可以看到每次迭代的开销从梯度下降的 O(n) 降到了常数 O(1)。因为随机梯度 ∇fi(x) 是对梯度 ∇f(x) 的无偏估计:
\mathbb{E}_i \nabla f_i(\boldsymbol{x}) = \frac{1}{n} \sum_{i = 1}^n \nabla f_i(\boldsymbol{x}) = \nabla f(\boldsymbol{x}).
平均上来说随机梯度是一个对梯度很好的估计。同梯度下降一样,如果选取合适的学习率,平均上每次迭代可以下降目标函数值。
一句话:随机梯度下降的更新轨迹相对于梯度下降更加曲折。因为加入的噪音(实际中,它来自样本的噪音)使得梯度的准确度下降,所以在使用同样的超参数的情况下,随机梯度下降收敛到的值相对梯度下降来说离最优值更远。但因为随机梯度下降每一次迭代的计算比梯度下降更加简单,在同样运行时间下,随机梯度下降可以进行更多次的自变量迭代,它最终得到的解的质量可能会比梯度下降更优。
2.3 小批量随机梯度下降法
- 当批量大小为 1 时,该算法即随机梯度下降;当批量大小等于训练数据样本数时,该算法即梯度下降。
- 当批量较小时,每次迭代中使用的样本少,这会导致并行处理和内存使用效率变低。这使得在计算同样数目样本的情况下比使用更大批量时所花时间更多。
- 当批量较大时,每个小批量梯度里可能含有更多的冗余信息,且在处理了同样多样本的情况下,它比批量较小的情况下对自变量的迭代次数更少,这两个因素共同导致了它靠近解的速度更慢。
- 可以通过调整的批量大小来权衡计算效率和训练误差下降速度。-
每一轮迭代里我们随机采样多个样本来组成一个小批量(mini-batch),然后对它计算梯度。这个算法被称为小批量随机梯度下降(mini-batch stochastic gradient descent)。通过重复采样(sampling with replacement)或者不重复采样(sampling without replacement)得到一个小批量中的各个样本。
\boldsymbol{g}_t \leftarrow \nabla f_{\mathcal{B}_t}(\boldsymbol{x}_{t-1}) = \frac{1}{|\mathcal{B}_t|} \sum_{i \in \mathcal{B}_t}\nabla f_i(\boldsymbol{x}_{t-1})
3 动量法
diss: 若目标函数在竖直方向(x2 轴方向)比在水平方向(x1 轴方向)的斜率的绝对值更大,当给定学习率,梯度下降迭代自变量时会使自变量在竖直方向比在水平方向移动幅度更大。因此一个较小的学习率从而避免自变量在竖直方向上越过目标函数最优解,造成了图中自变量在水平方向上朝最优解移动较慢。
动量法的提出是为了应对梯度下降的上述问题。在时间步 0,动量法创建速度变量 \boldsymbol{v}_0\in\mathbb{R}^d,并将其元素初始化成 0。在时间步 t>0,动量法对每次迭代的步骤做如下修改:
\begin{split}\begin{aligned} \boldsymbol{v}_t &\leftarrow \gamma \boldsymbol{v}_{t-1} + \eta_t \boldsymbol{g}_t \\ \boldsymbol{x}_t &\leftarrow \boldsymbol{x}_{t-1} - \boldsymbol{v}_t, \end{aligned}\end{split}
其中,动量超参数 γ 满足 0≤γ<1。当 γ=0 时,动量法等价于小批量随机梯度下降。由图可以看出,动量法在竖直方向上的移动更加平滑,且在水平方向上更快逼近最优解。
3.1 指数加权平均(EMA)
给定超参数 γ 且 0≤γ<1,当前时刻t 的变量 y_t 是上一时刻 t−1 的变量y_{t−1} 和当前时刻另一变量 x_t 的线性组合:
y_t = \gamma y_{t-1} + (1-\gamma) x_t.
对 y_t 展开:
\begin{split}\begin{aligned} y_t &= (1-\gamma) x_t + \gamma y_{t-1}\\ &= (1-\gamma)x_t + (1-\gamma) \cdot \gamma x_{t-1} + \gamma^2y_{t-2}\\ &= (1-\gamma)x_t + (1-\gamma) \cdot \gamma x_{t-1} + (1-\gamma) \cdot \gamma^2x_{t-2} + \gamma^3y_{t-3}\\ &\ldots \end{aligned}\end{split}
由于:
\lim_{n \rightarrow \infty} \left(1-\frac{1}{n}\right)^n = \exp(-1) \approx 0.3679,
令 n=1/(1−γ),那么有 (1−1/n)^n=γ^{1/(1−γ)}。所以当 γ→1 时,γ^{1/(1−γ)}=exp(−1)。例如 0.95^{20}=0.358≈exp(−1)。如果把 exp(−1) 当做一个比较小的数,我们可以在近似中忽略所有含 γ^{1/(1−γ)} 和比 γ^{1/(1−γ)} 更高阶的系数的项。例如,当 γ=0.95 时,y_t \approx 0.05 \sum_{i=0}^{19} 0.95^i x_{t-i}.
因此,在实际中,我们常常将 y 看作是对最近 1/(1−γ) 个时刻的 x 值的加权平均。例如,当 γ=0.95 时,y 可以被看作是对最近 20 个时刻的 x 值的加权平均;当 γ=0.9 时,y 可以看作是对最近 10 个时刻的 x 值的加权平均。且离当前时刻越近的 x 值获得的权重越大(越接近 1)
3.2 由指数加权移动平均理解动量法
现在,我们对动量法的速度变量做变形:
\boldsymbol{v}_t \leftarrow \gamma \boldsymbol{v}_{t-1} + (1 - \gamma) \left(\frac{\eta_t}{1 - \gamma} \boldsymbol{g}_t\right).
速度变量 v_t 实际上对序列 {η_{t−i}g_{t−i}/(1−γ):i=0,…,1/(1−γ)−1} 做了指数加权移动平均。换句话说,相比于小批量随机梯度下降,动量法在每个时间步的自变量更新量近似于将前者对应的最近 1/(1−γ) 个时间步的更新量做了指数加权移动平均后再除以 1−γ。
所以动量法中,自变量在各个方向上的移动幅度不仅取决当前梯度,还取决过去各个梯度在各个方向上是否一致。
例如:若梯度在水平方向上为正(向右)、而在竖直方向上时正(向上)时负(向下),自变量在水平方向的移动幅度逐渐增大,而在竖直方向的移动幅度逐渐减小。这样,我们就可以使用较大的学习率,从而使自变量向最优解更快移动。
- 动量法使用了指数加权移动平均的思想,其将过去时刻的梯度做了加权平均,且权重按时间指数衰减。
- 动量法使得相邻时间步之间的自变量更新在方向更加一致。
3.2 Nesterov
跟上面Momentum公式的唯一区别在于,,每次走之前,我们先用一个棍子往前探一探,这根棍子探到的位置就是L(θ_{n−1}−αΔθ_{n−1}),然后我们求解此处的梯度:如果梯度大,我们迈一大步,反之,迈一小步。

在原始形式中,Nesterov Accelerated Gradient(NAG)算法相对于Momentum的改进在于,以“向前看”看到的梯度而不是当前位置梯度去更新。经过变换之后的等效形式中,NAG算法相对于Momentum多了一个本次梯度相对上次梯度的变化量,这个变化量本质上是对目标函数二阶导的近似。由于利用了二阶导的信息,NAG算法才会比Momentum具有更快的收敛速度。
4 Adagrad
- diss: 梯度下降、(小批量)随机梯度下降、使用动量法,目标函数自变量的每一个元素在相同时刻都使用同一个学习率来自我迭代。当 x_1 和 x_2 的梯度值有较大差别时,我们需要选择足够小的学习率使得自变量在梯度值较大的维度上不发散。
- 动量法依赖指数加权移动平均使得自变量的更新方向更加一致,从而降低发散的可能。
- Adagrad 根据自变量在每个维度的梯度值的大小来调整各个维度上的学习率,从而避免统一的学习率难以适应所有维度的问题。
Adagrad 的算法会使用一个小批量随机梯度按元素平方的累加变量 s∈R_d。在时间步 0,adagrad 将 s_0中每个元素初始化为 0。在每次迭代中,首先将梯度 g_t 按元素平方后累加到变量 s_t:
\boldsymbol{s}_t \leftarrow \boldsymbol{s}_t + \boldsymbol{g}_t \odot \boldsymbol{g}_t,
再将目标函数自变量中每个元素的学习率通过按元素运算重新调整一下:
\boldsymbol{x}_t \leftarrow \boldsymbol{x}_{t-1} - \frac{\eta_t}{\sqrt{\boldsymbol{s}_t + \epsilon}} \odot \boldsymbol{g}_t,
其中 η_t 是学习率且一般为常数,ϵ 是为了维持数值稳定性而添加的常数,例如 10^{−6}。这里开方、除法和乘法的运算都是按元素进行的。这些按元素运算使得目标函数自变量中每个元素都分别拥有自己的学习率。
小批量随机梯度按元素平方的累加变量 s 出现在学习率的分母项中。
因此,如果目标函数有关自变量中某个元素的偏导数一直都较大,那么就让该元素的学习率下降快一点(稳定一些);反之,如果目标函数有关自变量中某个元素的偏导数一直都较小,那么就让该元素的学习率下降慢一点(加速)。
由于 s 一直在累加按元素平方的梯度,自变量中每个元素的学习率在迭代过程中一直在降低(或不变)。所以,当学习率在迭代早期降得较快且当前解依然不佳时,Adagrad 在迭代后期由于学习率过小,可能较难找到一个有用的解。
总结:
1. Adagrad 在迭代过程中不断调整学习率,并让目标函数自变量中每个元素都分别拥有自己的学习率。
2. 使用 Adagrad 时,自变量中每个元素的学习率在迭代过程中一直在降低(或不变)。
5 RMSprop
Diss: Adagrad s 一直在累加按元素平方的小批量随机梯度,目标函数自变量每个元素的学习率在迭代过程中一直在降低(或不变)。所以,当学习率在迭代早期降得较快且当前解依然不佳时,Adagrad 在迭代后期由于学习率过小,可能较难找到一个有用的解。
在RMSProp 将过去时间步里梯度按元素平方做指数加权移动平均,给定超参数 γ 且 0≤γ<1,RMSProp 在时间步 t>0 里计算
\boldsymbol{s}_t \leftarrow \gamma \boldsymbol{s}_{t-1} + (1 - \gamma) \boldsymbol{g}_t \odot \boldsymbol{g}_t.
和 Adagrad 一样,RMSProp 将目标函数自变量中每个元素的学习率通过按元素运算重新调整一下,然后更新自变量。
\boldsymbol{x}_t \leftarrow \boldsymbol{x}_{t-1} - \frac{\eta_t}{\sqrt{\boldsymbol{s}_t + \epsilon}} \odot \boldsymbol{g}_t,
其中 η_t 是学习率,ϵ 是为了维持数值稳定性而添加的常数,例如 10^{−6}
因为 RMSProp 的状态变量是对平方项 g_t⊙g_t 的指数加权移动平均,因此可以看作是最近 1/(1−γ) 个时刻的梯度平方项的加权平均,这样自变量每个元素的学习率在迭代过程中避免了“直降不升”的问题。
总结:
- RMSProp 和 Adagrad 的不同在于,RMSProp 使用了小批量随机梯度按元素平方的指数加权移动平均变量来调整学习率。
- 理解指数加权移动平均有助于我们调节 RMSProp 算法中的超参数,例如 γ。
6 Adadelta
没有学习率
Adadelta 算法也像 RMSProp 一样,使用了小批量随机梯度按元素平方的指数加权移动平均变量 s,它的时间步 0 时被初始化为 0。 给定超参数 ρ 且 0≤ρ<1(对应 RMSProp 中的 γ),在时间步 t>0,同 RMSPro 一样计算:
\boldsymbol{s}_t \leftarrow \rho \boldsymbol{s}_{t-1} + (1 - \rho) \boldsymbol{g}_t \odot \boldsymbol{g}_t.
不同的在于 Adadelta 算法还维护一个额外的状态变量 \Delta\boldsymbol{x}\in\mathbb{R}^d,其元素同样在时间步 0 时被初始化为 0。然后使用它来计算自变量的变化量:
\boldsymbol{g}_t' \leftarrow \sqrt{\frac{\Delta\boldsymbol{x}_{t-1} + \epsilon}{\boldsymbol{s}_t + \epsilon}} \odot \boldsymbol{g}_t,
接着更新自变量:
\boldsymbol{x}_t \leftarrow \boldsymbol{x}_{t-1} - \boldsymbol{g}'_t.
最后,我们使用 Δx 来记录 g′ 按元素平方的指数加权移动平均:
\Delta\boldsymbol{x}_t \leftarrow \rho \Delta\boldsymbol{x}_{t-1} + (1 - \rho) \boldsymbol{g}'_t \odot \boldsymbol{g}'_t.
Adadelta 跟 RMSProp 不同的地方在于使用 Δx_t 来替代了超参数 ηt,因此它的主要优势在于不需要手动选取学习率。
总结:
- AdaDelta 没有学习率参数,它通过使用自变量更新量平方的指数加权移动平均来替代学习率。
- 两个EMA
7 Adam
Diss: RMSProp 的EMA对0拖后腿 轮启动问题。
将 v_t 再除以 1−β^{t}_1,从而使得过去各时刻小批量随机梯度权值之和为 1。(0≤β1<1,算法作者建议设为 0.9)
\boldsymbol{v}_t \leftarrow \beta_1 \boldsymbol{v}_{t-1} + (1 - \beta_1) \boldsymbol{g}_t.
偏差修正:
\boldsymbol{v}'_t \leftarrow \frac{\boldsymbol{v}_t}{1 - \beta_1^t}.
接下来和 RMSProp 中一样,给定超参数 β2 且满足 0≤β2<1(算法作者建议设为 0.999),更新状态变量 s:
\boldsymbol{s}_t \leftarrow \beta_2 \boldsymbol{s}_{t-1} + (1 - \beta_2) \boldsymbol{g}_t \odot \boldsymbol{g}_t.
且同样做偏差修正:
\boldsymbol{s}'_t \leftarrow \frac{\boldsymbol{s}_t}{1 - \beta_2^t}.
最后使用修正后的变量 v_t' 和 s_t' 来更新自变量:
\boldsymbol{x}_{t} \leftarrow \boldsymbol{x}_{t-1} - \frac{\eta}{\sqrt{\boldsymbol{s}_{t}'+\epsilon}}\odot\boldsymbol{v}_{t}'
总结:
修正:t小——>v放大修正,t大——>v正常
8 牛顿法
牛顿法不仅使用了一阶导信息,同时还利用了二阶导来更新参数,其形式化的公式如下:
\theta_n:=\theta_{n-1}-\alpha\frac{L^{'}_{n-1}}{L^{''}_{n-1}}
回顾之前的θ_n=θ_{n−1}+Δθ,我们将损失函数在θn−1处进行二阶泰勒展开:
L(\theta_n)=L(\theta_{n-1}+\Delta\theta)\approx L(\theta_{n-1})+L^{'}(\theta_{n-1})\Delta\theta+\frac{L^{''}(\theta_{n-1})\Delta\theta^2}{2}
要使L(θ_n)
\Delta\theta = -\frac{L^{'}_{n-1}}{L^{''}_{n-1}}
也即牛顿法的迭代公式,拓展到高维数据,二阶导变为Hession矩阵,上式变为:
\Delta\theta = -H^{-1}L^{'}_{n-1}
直观上,我们可以这样理解:我们要求一个函数的极值,假设只有一个全局最优值,我们需要求得其导数为0的地方,我们把下图想成是损失函数的导数的图像f(x),那么:
k=\tan \theta = f'(x_0)=\frac{f(x_0)}{x_0-x_1}\rightarrow x_1= x_0 - \frac{f(x_0)}{f'(x_0)}
我们一直这样做切线,最终x_n将逼近与f′(x)的0点,对于原函数而言,即\Delta\theta = -\frac{L^{'}_{n-1}}{L^{''}_{n-1}}。

牛顿法具有二阶收敛性,每一轮迭代会让误差的数量级呈平方衰减。即在某一迭代中误差的数量级为0.01,则下一次迭代误差为0.0001,再下一次为0.00000001。收敛速度快,但是大规模数据时,Hession矩阵的计算与存储将是性能的瓶颈所在。
为此提出了一些算法,用来近似逼近这个Hession矩阵,最著名的有L-BFGS,优于BFGS,可适用于并行计算从而大大提高效率
牛顿法的优缺点总结:
优点:二阶收敛,收敛速度快;
缺点:牛顿法是一种迭代算法,每一步都需要求解目标函数的Hessian矩阵的逆矩阵,计算比较复杂。
9 拟牛顿法
拟牛顿法的本质思想是改善牛顿法每次需要求解复杂的Hessian矩阵的逆矩阵的缺陷,它使用正定矩阵来近似Hessian矩阵的逆,从而简化了运算的复杂度。
拟牛顿法的基本思想如下:
1.首先构造目标函数在当前迭代x_k的二次模型:
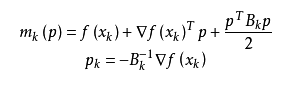
这里Bk是一个对称正定矩阵,于是我们取这个二次模型的最优解作为搜索方向,并且得到新的迭代点:

其中我们要求步长a_k 满足Wolfe条件。这样的迭代与牛顿法类似,区别就在于用近似的Hesse矩阵B_k 代替真实的Hesse矩阵。所以拟牛顿法最关键的地方就是每一步迭代中矩阵B_k 的更新。现在假设得到一个新的迭代x_k+1,并得到一个新的二次模型:

我们尽可能地利用上一步的信息来选取B_k。具体地,我们要求

从而得到
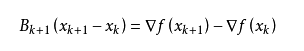
这个公式被称为割线方程。常用的拟牛顿法有DFP算法和BFGS算法。


参考
[1]最全的机器学习中的优化算法介绍
[2] Gluon
[3] 各种优化方法总结