---------------
Nickier
2018-12-10
---------------
RNA
1.视频
测序流程:提取总RNA--去除rRNA--逆转录cDNA--打断--加接头--上机测序
因为95%+都是rRNA
建库
因此mRNA富集
Y型接头(adapter)
建库会评级,比如A B C级,如RNA质量的评估
既然是mRNA测序,大多数测序的读长(reads)应该富集在cDNA区域,可以通过IGV可视化
2.文献阅读
流程
Q30,
Q20
GC含量
3.数据分析流程
原始数据(raw data),fq
数据质控(fastqc multiqc),html
比对(hisat),bam
问题:比对不能用bwa或者bowtie?RNA-seq和wes的序列有什么区别?
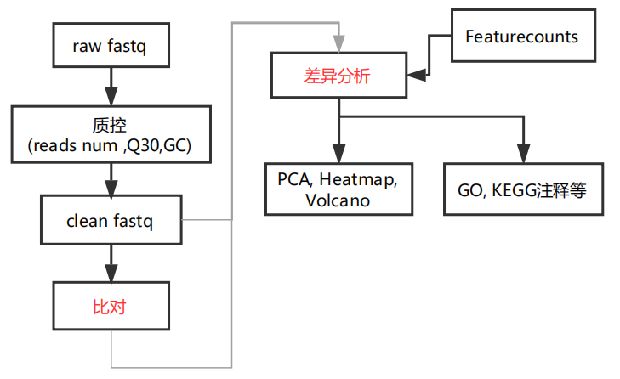
4.实战流程
主要流程有:
准备工作:下载数据:进入ncbi下载,下载SRA号,SRR_Acc_list.txt,上传到服务器
0.prepare work(install software)
1.data download(sra-->fq)
2.QC
3.filter and remove adapter
4.alignment
5.counts
6.DEG(R)
0.软件安装
0.1安装conda
Conda 是一个开源的软件包管理系统和环境管理系统,用于安装多个版本的软件包及其依赖关系,并在它们之间轻松切换。
搜索 miniconda 清华,拿到miniconda的linux版本,然后再到服务器下载
# 在linux在使用以下命令下载miniconda
wget -c https://mirrors.tuna.tsinghua.edu.cn/anaconda/miniconda/Miniconda3-latest-Linux-x86_64.sh
# 载入激活
bash Miniconda3-latest-Linux-x86_64.sh
source ~/.bashrc
# 添加镜像
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/pkgs/free
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/conda-forge
conda config --add channels https://mirrors.tuna.tsinghua.edu.cn/anaconda/cloud/bioconda
conda config --set show_channel_urls yes
0.2 使用conda安装其他软件
可能需要安装的软件:
质控:fastqc , multiqc, trimmomatic, ,trim-galore
比对:star, hisat2, bowtie2, tophat, bwa, subread,samtools
计数:htseq, bedtools, deeptools, salmon
# 创建一个rna工作环境
conda create -n rna python=2
# 激活环境
source activate rna
# 安装软件,安装之前先search一下,conda search software
conda install -y sra-tools
conda install -y fastqc
conda install -y multiqc
conda install -y samtools
conda install -y trim-galore
conda install -y hisat2
conda install -y subread
1. data download(sra---->fq)
1.1 下载sra的id号
首先在文献中找到id号,如GSE63310,在NCBI上的地址是https://www.ncbi.nlm.nih.gov/gds/?term=GSE63310。然后找到其SRA号SRP049824,进入,下载得到SRR_Acc_List.txt,也可以把SRR号的id复制到txt文本中。最后把SRR_Acc_List.txt上传到服务器。
1.2 用sra-tools的命令prefetch下载数据
# 首先创建一个工作目录rnaseq,进入
mkdir ~/rnaseq
cd ~/rnaseq
# 用sra-tools的命令prefetch下载数据
cat SRR_Acc_List.txt | while read id; do (prefetch ${id} &);done
# 因为数据比较大,练习的时候可以用服务器上已经下载好的数据
# 把上面的进程kill掉
ps -ef | grep prefetch | awk '{print $2}' | while read id; do kill ${id}; done1.3 用下载好的数据转换格式sra-->fq
# 如下载好的数据保存在了~/rnaseq/raw/
cd ~/rnaseq/raw/
ls ~/rnaseq/raw/*.sra | while read id; do(nohup fastq-dump --gzip --split-3 -O ./ ${id} &);done
# 原始数据是双端测序结果,fastq-dump配合--split-3参数,一个样本被拆分成两个fastq文件,此时~/rnaseq/raw/下面就生成了fq文件,如SRR1039508_1.fastq.gz SRR1039508_2.fastq.gz
2. QC
对现有的fq文件进行质控,生成html质控报告,质控用到的软件是fastqc和multiqc,后者生成的是整合后的质控报告
# 在~/rnaseq/目录创建一个文件夹qc
mkdir qc
cd qc
# 跑fastqc,生成了html质控报告文件
ls ~/rnaseq/raw/*gz | xargs fastqc -t 10 -o ./
# 跑multiqc,对fastqc的结果进行整合,生成multiqc_report.html
multiqc ./
# 得到质控报告后就下载到本地查看,看看是否需要进行过滤和去接头,即下一步
3. filter and remove adapter
3.1 小样本比对
在样本比较少的情况下,如现在只有SRR1039508_1.fastq.gz SRR1039508_2.fastq.gz两个样本,可以用比较简单的办法过滤,样本量比较大时,就用脚本。
# 在~/rnaseq/目录下创建文件夹clean
mkdir clean
cd clean
# 过滤掉质量差的reads和去adapter,参数说明:
# nohup 在后台运行
# --phred33 质量值体系为33
# -q 25 质量值低于25过滤掉
# -e 0.1 允许的最大错误率(错误数除以匹配的长度)0.1
# --length 36 设定输出reads长度阈值为36,小于设定值36会被抛弃
# --stringency 3 设定前后adapter重叠的碱基数为3,大于3的会被过滤
# --paired 对于双端测序结果,一对reads中,如果有一个被剔除,那么另一个会被同样抛弃,而不管是否达到标准
# nohup 将命令挂在后台的意思
nohup trim_galore --phred33 -q 25 -e 0.1 --length 36 --stringency 3 --paired -o ~/rnaseq/clean/ ~/rnaseq/qc/SRR1039508_1.fastq.gz ~/rnaseq/qc/SRR1039508_2.fastq.gz &
3.2 如果是样本量比较大,可以使用脚本
# 首先把样本名称存入到文本config中
ls ~/rnaseq/clean/*_1.fastq.gz >1
ls ~/rnaseq/clean/*_2.fastq.gz >2
paste 1 2 > config
# 然后建一个qc.sh
touch qc.sh
# 写入以下内容
source activate rna
dir=~/rnaseq/clean/
bin_trim_galore=trim_galore
cat config |while read id
do
arr=(${id})
fq1=${arr[0]}
fq2=${arr[1]}
nohup $bin_trim_galore -q 25 --phred33 --length 36 --stringency 3 --paired -o $dir $fq1 $fq2 &
done
# 最后运行
bash qc.sh
# 运行之后会生成SRR1039508_1_val_1.fq.gz和SRR1039508_2_val_2.fq.gz
# 可以再做一次QC4. alignment
4.1 对于RNAseq,比对的时候要用hisat2或subread,不能用bowtie2或者bwa
# 首先在~/rnaseq/目录下创建文件夹align
mkdir align
cd align
# 然后比对,用hisat2,比对结果可以输出到sam文件,再转成bam文件,也可以用samtools直接生成bam文件。
# 参数说明:
# -p 10 调用10个线程
# -x /public/reference/index/hisat/hg38/genome 参考基因组路径
#
hisat2 -p 10 -x /public/reference/index/hisat/hg38/genome -1 ~/rnaseq/clean/SRR1039508_1_val_1.fq -2 ~/rnaseq/clean/SRR1039508_2_val_2.fq | samtools sort -@ 5 -o SRR1039508.sort.bam -4.2 同样的,样本较多时,可以使用循环命令来比对
ls ~/rnaseq/clean/*gz|cut -d"_" -f 1|sort -u|while read id
do
ls -lh ${id}_1_val_1.fq.gz ${id}_2_val_2.fq.gz
hisat2 -p 10 -x /public/reference/index/hisat/hg38/genome -1 ${id}_1_val_1.fq.gz -2 ${id}_2_val_2.fq.gz -S ${id}.hisat.sam
done
# 生成了hisat.sam文件,再用samtools将sam文件转成bam文件
ls *.sam|while read id ;do (samtools sort -O bam -@ 5 -o $(basename ${id} ".sam").bam ${id});done
4.3 为bam文件建索引
ls *.bam |xargs -i samtools index {}4.4 可以对比对后的结果进行统计
ls *.bam |while read id ;do ( nohup samtools flagstat -@ 1 $id > $(basename ${id} ".bam").flagstat & );done5. counts
对转录组进行定量,基于基因水平的定量,用subread的featureCounts来获取每个基因上的count数。所谓count数,个为根据不同比对情况,将reads分配到各个基因上。counts之后就得到了表达矩阵了,即all.id.txt文件
# 首先,把gtf文件赋值给变量gtf
gtf="/public/reference/gtf/gencode/gencode.v25.annotation.gtf.gz"
# 然后运行featureCounts命令
# 参数说明:
# -T 5 指5个线程
# -p 这个参数是针对paired-end数据
# -t exon 从注释文件gtf提取exon信息,默认将exon作为一个feature
# -g gene_id 从注释文件gtf中提取Meta-features信息用于read count,默认是gene_id
# -a 输入GTF/GFF基因组注释文件
# -o 输出
featureCounts -T 5 -p -t exon -g gene_id -a $gtf -o all.id.txt *.bam 1>counts.id.log 2>&1 &
# 最后就得到了all.id.txt文件,表达矩阵,下一步就是导入到R中进行分析了6.DEG
RStudio
差异分析之前需要首先对转录组上游分析得到的文件 all.id.txt 进行一定程度的检查,代码如:
rm(list = ls())
options(stringsAsFactors = F)
a=read.table('all.id.txt',header = T)
tmp=a[1:14,1:7]
meta=a[,1:6]
exprSet=a[,7:ncol(a)]
colnames(exprSet)
a2=exprSet[,'SRR1039516.hisat.bam']
library(airway)
data(airway)
exprSet=assay(airway)
colnames(exprSet)
a1=exprSet[,'SRR1039516']
group_list=colData(airway)[,3]
a2=data.frame(id=meta[,1],a2=a2)
a1=data.frame(id=names(a1),a1=as.numeric(a1))
library(stringr)
a2$id <- str_split(a2$id,'\\.',simplify = T)[,1]
tmp=merge(a1,a2,by='id')
png('tmp.png')
plot(tmp[,2:3])
dev.off()
library(corrplot)
png('cor.png')
corrplot(cor(log2(exprSet+1)))
dev.off()
library(pheatmap)
png('heatmap.png')
m=cor(log2(exprSet+1))
pheatmap(scale(cor(log2(exprSet+1))))
dev.off()