文章介绍
1| 标题:MetaCHIP: community-level horizontal gene transfer identification through the combination of best-match and phylogenetic approaches[1]
2| 热心肠解读:[2]
- MetaCHIP主要通过BLASTN搜索最佳匹配、进化树构建等两种方法分析水平基因转移;
- 该流程整合了宏基因组序列组装、分箱、基因预测、序列比对和进化树分析的多个生物信息学工具;
- 对仿真序列的测试表明该工具可以得出近期和非近期的基因水平转移事件;
- 应用于人肠道和土壤菌群等真实实验数据时,与已发表研究中的结果吻合,同时还能额外发现新的转移事件;
- 基因转移后发生大程度突变、序列组装、分箱结果等可能限制该方法的运用。
软件介绍及下载
1| 软件介绍:
MetaCHIP是一款分析群体水平基因转移的工具流程,基于Python开发,并且利用一系列第三方工具包和R语言包。关于该包的详细信息在github网站[3],包括案例数据,教程及涉及的软件
2| 软件下载:
# 我的是基于conda环境下载的,但是如果安装了pip软件,应该也可以
$ source activate qiime2-2018.6
$ pip install MetaCHIP
$ MetaCHIP -h # 测试软件是否能正常运行
...::: MetaCHIP v1.1.10 :::...
HGT detection modules:
PI -> Prepare Input files
BM -> Best-Match approach
PG -> PhyloGenetic approach
# for command specific help
MetaCHIP PI -h
MetaCHIP BM -h
MetaCHIP PG -h
备注:MetaCHIP的输入文件需要:
- 包含fasta格式的序列文件(如构建好的bin)
- 以及一个文本文件(.txt格式),需包含物种分类信息,并且这里要求序列ID信息不能超过22个字符
软件使用
MetaCHIP软件包含3个模块(如上方的帮助信息所示),有PI、BM和PG模块,下面分别详细介绍这三个模块的使用[4]
1| PI模块
$ MetaCHIP PI -h # 获得PI模块的使用参数
usage: MetaCHIP PI [-h] -i I [-taxon TAXON] -p P [-r R] [-g G] [-x X]
[-grouping_only] [-nonmeta] [-noblast] [-t T] [-qsub]
[-force] [-quiet]
Prepare input files
optional arguments:
-h, --help show this help message and exit
-i I input genome folder # 文件夹名称,里面含序列文件(如一些构建的bin文件)
-taxon TAXON taxonomic classification # 物种分类信息文件
-p P output prefix # 输出文件的前缀
-r R grouping rank # 物种分类的层级,门、纲、目、科、属等
-g G grouping file # 分组文件
-x X file extension # 文件拓展名
-grouping_only run grouping only, deactivate Prodigal and Blastn
-nonmeta annotate Non-metagenome-assembled genomes (Non-MAGs)
-noblast not run all-vs-all blastn
-t T number of threads, default: 1
-qsub run blastn with job scripts, only for HPC users
-force overwrite previous results
-quiet not report progress
1.1| 模块解释
PI 模块的主要目的就是为了将数据文件:序列文件和分组信息文件整合在一起。建议使用GTDBTk数据库作为输入序列文件的物种分类数据库。
1.2| 示例命令
# grouping input genomes at provided levels according to taxonomic classifications
$ MetaCHIP PI -i soil_bins -x fa -taxon GTDB_op.tsv -r c -p Soil -t 6
$ MetaCHIP PI -i soil_bins -x fa -taxon GTDB_op.tsv -r o -p Soil -t 6 -grouping_only
$ MetaCHIP PI -i soil_bins -x fa -taxon GTDB_op.tsv -r f -p Soil -t 6 -grouping_only
# run with customized grouping profile
$ MetaCHIP PI -i soil_bins -x fa -g customized_grouping.txt -p Soil -t 6
备注:输出文件
- 组合结果输出到[prefix]grouping_[taxon_rank][group_num].txt
- 基因检出结果存储为GenBank和fasta格式
- 根据输入序列文件产生一个SCG蛋白tree文件
- 条形图显示了提供特定的分类学等级的每组中基因组的数量
- blast结果
2| BM模块
$ MetaCHIP BM -h
usage: MetaCHIP BM [-h] -p P [-r R] [-g G] [-cov COV] [-al AL] [-flk FLK]
[-ip IP] [-ei EI] [-t T] [-plot_iden] [-NoEbCheck] [-force]
[-quiet] [-tmp]
Best-match approach
optional arguments:
-h, --help show this help message and exit
-p P output prefix #上一步输出文件的前缀,如Soil
-r R grouping rank # 提供分类等级
-g G grouping file # 提供分组文件
-cov COV coverage cutoff, default: 75 # 覆盖度
-al AL alignment length cutoff, default: 200
-flk FLK the length of flanking sequences to plot (Kbp), default: 10
-ip IP identity percentile cutoff, default: 90
-ei EI end match identity cutoff, default: 95
-t T number of threads, default: 1
-plot_iden plot identity distribution
-NoEbCheck disable end break and contig match check for fast processing,
not recommend for metagenome-assembled genomes (MAGs)
-force overwrite previous results
-quiet Do not report progress
-tmp keep temporary files
2.1| 模块解释
通过最佳匹配方法进行基因水平转移候选预测,以及对其侧翼区域进行作图输出,文件格式如下:
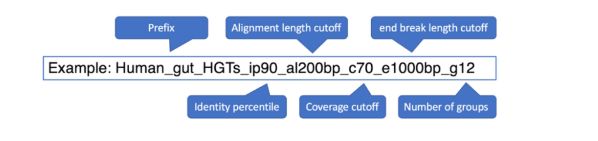
2.2| 示例命令
$ MetaCHIP BM -p Soil -r c -t 6
# run with customized grouping profile
$ MetaCHIP BM -p Soil -g customized_grouping.txt -t 6
备注:输出文件
- BM方法得到的水平转移基因输出到文件HGT_candidates_BM.txt.
- 对应基因的核酸和氨基酸序列分别输出到HGT_candidates_BM_nc.fasta和HGT_candidates_BM_aa.fasta
3| PG模块
$ MetaCHIP PG -h
usage: MetaCHIP PG [-h] -p P [-r R] [-g G] [-cov COV] [-al AL] [-flk FLK]
[-ip IP] [-ei EI] [-t T] [-force] [-quiet]
Phylogenetic approach
optional arguments:
-h, --help show this help message and exit
-p P output prefix # 输出文件前缀
-r R grouping rank
-g G grouping file
-cov COV coverage cutoff, default: 75
-al AL alignment length cutoff, default: 200
-flk FLK the length of flanking sequences to plot (Kbp), default: 10
-ip IP identity percentile, default: 90
-ei EI end match identity cutoff, default: 95
-t T number of threads, default: 1
-force overwrite previous results
-quiet Do not report progress
3.1| 模块解释
PG:phylogenetic approach, 该模块主要目的就是构建系统发育树
3.2| 示例命令
$ MetaCHIP PG -p NorthSea -r c -t 6
# run with customized grouping profile
$ MetaCHIP PG -p NorthSea -g customized_grouping.txt -t 6
备注:输出文件
候选水平转移基因(horizontal gene transfer, HGT)通过PG方法进一步验证并输出到和BM同一个文件夹下。
- HGT_candidates_PG.txt:BM预测得到的水平转移基因,PG提供了额外的信息。
- HGT_candidates_PG_validated.txt:仅由PG方法验证到的HGT
- HGT_candidates_PG_nc.fasta:通过PG方法验证得到的HGT核酸序列
- HGT_candidates_PG_aa.fasta:通过PG方法验证得到的HGT氨基酸序列
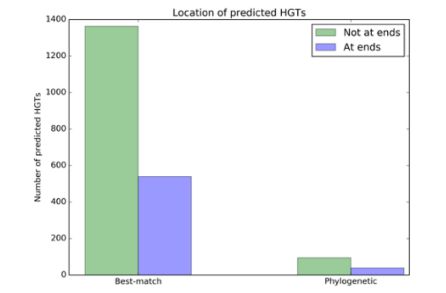
- [prefix]_plot_at_ends_stat.png:通过BM和PG预测得到HGT位置统计
- [prefix]_plot_HGT_identities_BM.png:BM方法预测得到的HGT的identity分布
[图片上传失败...(image-90f350-1553326730141)]
- [prefix]_plot_HGT_identities_PG.png:通过PG方法验证到的HGT的identity分布
[图片上传失败...(image-47a14e-1553326730141)]
- [prefix]_plot_circos_PG.png:不同组之间的基因流向。供体和受体之间通过形变带进行连接,变形带的宽度表示相关的HGT的数目。

参考
-
原文 ↩
-
热心肠解读 ↩
-
MetaCHIP-github ↩
-
MetaCHIP-manual ↩