
# findMotifsGenome.pl: 在基因组区域中寻找富集Motifs
HOMER 最初设计的目的用于ChIP-Seq peaks 中寻找富集motifs 。
#命令
findMotifsGenome.pl #1. 设定寻找motif 的区域大小 (-size # or -size given, default: 200)
如果想在提供的peak中寻找motifs,使用参数-size given。然而,对于转录因子peaks,大多数motifs 被发现位于peak 中心 +/- 50-75 bp的范围内,所以最好根据peak 的大小将寻找motif的区域设为固定值。
#2. 输入文件格式
格式: HOMER peak/Positions 文件和BED 格式文件
HOMER peak文件有至少5列:
- Column1: Peak ID
- Column2: 染色体
- Column3: 起始位置
- Column4: 终止位置
- Column5: 链的方向(+/- or 0/1, where 0="+", 1="-")
BED 格式文件至少有6列:
- Column1: 染色体
- Column2: 起始位置
- Column3: 终止位置
- Column4: Peak ID
- Column5: not used
- Column6: 链的方向 (+/- or 0/1, where 0="+", 1="-")
Peak/Position 和 BED两种格式之间可以相互转换,使用Homer自带脚本:pos2bed.pl 或bed2pos.pl
#3. 自定义背景
因为HOMER 使用一个不同的motif 寻找算法,因此使用不同的背景会产生不同的结果。例如,如果将某种实验的peak与另一种实验peak相比较,可以再创建一个peak/BED文件(参数:"-bg
#4. findMotifsGenome.pl工作流程
4.1 确认peak/BED 文件
4.2 根据peak/BED 文件提取序列,过滤掉序列中N >70%的序列。
4.3 计算peak 对应序列GC/CpG含量
4.4 根据设定的大小准备背景序列
用于寻找motif 区域大小使用("-size <#>")设置。HOMER 一般选取基因TSS +/- 50kb区域分成设定大小;然后计算这些背景序列GC/CpG% 储存起来用于后续分析。
4.5 随机选择背景区域用于寻找motif
因为HOMER 使用一个不同的motif 寻找算法,它需要使用背景序列区域作为对照。默认情况下,HOMER 可能选择50000 或 peaks总数两倍的随机背景序列,可以使用参数-N <#>自定义。HOMER 会选择和目标数据一致GC 含量分布的序列作为背景序列。例如,目标序列是GC高含量的,那么背景序列也会如此。
设定-bg 自定义背景,
4.6 序列差异自动标准化
自动标准化是HOMER 用以移除由短寡聚序列引进的序列偏好性,主要用于消除某些特定基因组序列、实验误差和测序偏好引起的不平衡。HOMER 假定目标数据和背景序列在1-mers, 2-mers, 3-mers, etc上是没有差异的。短寡聚序列长度是通过参数-nlen <#>设定。一个例子,目标数据和背景序列中 A's是一样的;先计算目标序列中各种短寡聚序列的偏好性,然后调整每条背景序列的权重来标准化这些偏好性,当然权重矫正是按照较小的步长一步一步进行矫正。如果目标序列富含A,那么背景序列中富含A的序列权重高于A含量一般的序列。
4.7 检查已知motifs富集情况
HOMER 会检索已知 motifs 在目标序列和背景基因富集情况。结果输出到文件:knownResults.html
4.8 重头预测motif
默认情况,HOMER 寻找长度为 8, 10, 和12 bp的motifs ,可以通过-len <#,#,#>自定义。
5 findMotifsGenome.pl结果文件
- homerMotifs.motifs<#> : 对应各个长度的motif结果
- homerMotifs.all.motifs : 各个长度的motif结果合并到了一起
- motifFindingParameters.txt : 文件保存了程序运行参数
- knownResults.txt : 已知motif 的富集结果
- seq.autonorm.tsv : 短核苷酸自动矫正情况
- homerResults.html : 重新预测的motif 的富集结果
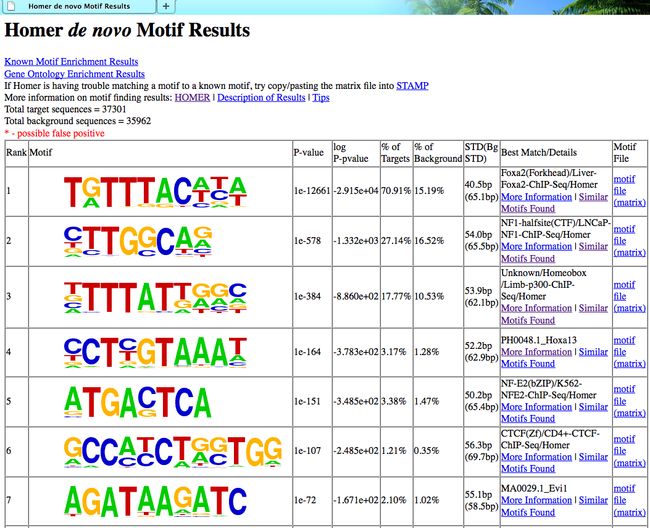
- homerResults/ directory: 对应homerResults.html中结果
- knownResults.html : 已知motif 的富集结果
- knownResults/ directory: 对应knownResults.html 中结果
#6 Interpreting motif finding results
#7 motif 寻找的一些重要参数
Masked vs. Unmasked Genome ("-mask" or hg18 vs. hg18r)
一般使用masked 版本Region Size ("-size <#>", "-size <#>,<#>", "-size given", default: 200)
-size -300,100:peak上游100bp,下游300bp区域。根据不同的实验数据选择。Motif length ("-len <#>" or "-len <#>,<#>,...", default 8,10,12)
如果要寻找长Motif ,建议先寻找短的Motif(<15bp);寻找长的Motif 耗时和占据大量计算机资源,建议减小寻找Motif 的区域,例如"-len 20 -size 50"。Mismatches allowed in global optimization phase ("-mis <#>", default: 2)
允许错配可以提升灵敏度,如果寻找12-15 bp Motif ,可以设置3-4bp的错配。Number of motifs to find ("-S <#>", default 25)
并不是越多越好。Normalize CpG% content instead of GC% content ("-cpg")
考虑到HOMER 可能卡在CGCGCGCG这样的motifs。Region level autonormalization ("-nlen <#>", default 3, "-nlen 0" to disable)
消除短寡聚核苷酸引入的不平衡。Motif level autonormalization (-olen <#>, default 0 i.e. disabled)
对Region level autonormalization参数的补充。User defined background regions ("-bg
")
自定义背景序列Hypergeometric enrichment scoring ("-h")
findMotifsGenome.pl默认使用二项式分布对motifs打分,这是因为背景序列远远多于目标序列时,运算比较快。当背景序列比较少的时候,建议使用超几何检验的方法。Find enrichment of individual oligos ("-oligo")
输出寡聚核苷酸富集情况到文件oligo.length.txtForce findMotifsGenome.pl to re-preparse genome for the given region size ("-preparse").
Only search for motifs on + strand ("-norevopp")
Search for RNA motifs ("-rna")
Mask motifs ("-mask
") Optimize motifs ("-opt
") Dump FASTA files ("-dumpFasta")
根据peak文件输出 target.fa 和 background.fa
#8. findMotifsGenome.pl使用实例:
8.1 数据包准备
$perl configureHomer.pl -list
$perl configureHomer.pl -install mm10
8.2 构建HOMER Peak/Positions 文件
#input.test.bed
#peakName #chromsome #startingPosition #endPosition #strand
1 chr2 5214158 5215219 +
2 chr2 8345384 8345769 +
3 chr2 8647810 8648265 +
4 chr2 8943836 8944187 +
5 chr2 10036538 10036796 +
6 chr3 12362628 12362865 +
7 chr3 13105367 13105590 +
8 chr3 15619314 15619600 +
9 chr3 19819943 19820193 +
10 chr3 22236595 22236910 +
8.3 运行程序
$ perl findMotifsGenome.pl input.test.bed mm10 /homerResult/ -size 200 -len 8,10,12
常用参数:
-bg:自定义背景序列
-size: 用于motif寻找得片段大小,默认200bp;-size given 设置片段大小为目标序列长度;越大需要得计算资源越多
-len:motif大小设置,默认8,10,12;越大需要得计算资源越多
-S:结果输出多少motifs, 默认25
-mis:motif错配碱基数,默认2bp
-norevopp:不进行反义链搜索motif
-nomotif:关闭重投预测motif
-rna: 输出RNA motif,使用RNA motif数据库
-h:使用超几何检验代替二项式分布
-N:用于motif寻找得背景序列数目,default=max(50k, 2x input);耗内存参数
参考:
Finding Enriched Motifs in Genomic Regions
ChIP-Seq 数据挖掘系列文章目录:
ChIP-Seq数据挖掘系列-1:Motif 分析(1)-HOMER 安装
ChIP-Seq数据挖掘系列-2: Motif 分析(2) - HOMER Motif 分析基本步骤
ChIP-Seq数据挖掘系列-3: Motif 分析(3) - 利用ChIP-Seq结果在基因组区域中寻找富集的Motifs
ChIP-Seq数据挖掘系列-4: liftOver - 基因组坐标在不同基因组注释版本间转换
ChIP-Seq数据挖掘系列-5.1: ngs.plot 可视化ChIP-Seq 数据
ChIP-Seq数据挖掘系列-5.2: ngs.plot 画图工具ngs.plot.r 和 replot.r 参数详解