首先最重要的参考链接:
- [karyoploteR]
- https://bernatgel.github.io/karyoploter_tutorial/Examples/EncodeEpigenetics/EncodeEpigenetics.html
其次我就是一个搬运工,希望给有需要的小伙伴,如有老哥看到侵权了,麻烦请私信我删除,谢谢!
番外在和小伙伴分享的时候,小伙伴也发了一个可以完成此功能的链接 trackViewer Vignette
- 由于我的傻吊服务器装不上包,后面需要linux的基本就是复制粘贴看了一遍过来的。。。 话说服务器装R包我是真的很无语
-
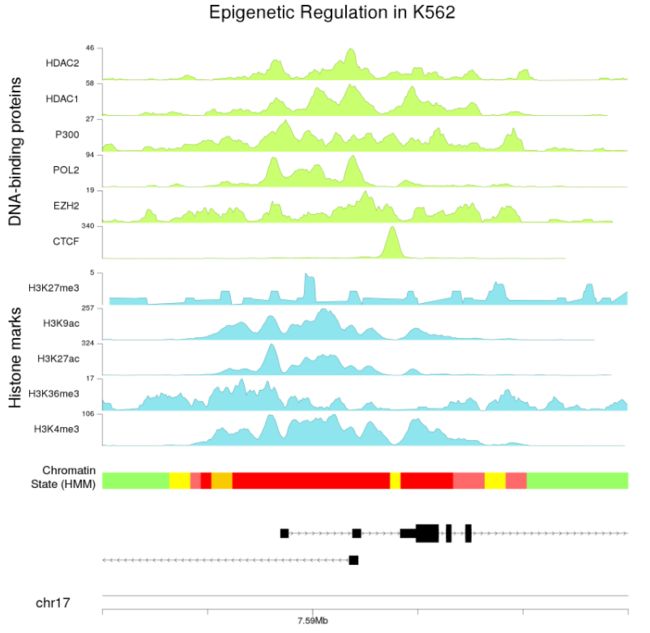
那么重要的是通过此文我们可以学会绘制出什么样的图呢?见下:
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("karyoploteR", version = "3.8")
## 哇,按照上面安装得到的是1.8.8版本的,然后会发现后面很多命令都没有。。
# 从新安装
devtools::install_github("bernatgel/karyoploteR") # karyoploteR_1.9.21版本
library(karyoploteR)
TP53.region <- toGRanges("chr17:7,564,422-7,602,719")
> TP53.region
GRanges object with 1 range and 0 metadata columns:
seqnames ranges strand
[1] chr17 7564422-7602719 *
-------
seqinfo: 1 sequence from an unspecified genome; no seqlengths
kp <- plotKaryotype(zoom = TP53.region)

- 使用
kpPlotGenes函数绘制上面展示区域内的基因。
BiocManager::install("TxDb.Hsapiens.UCSC.hg19.knownGene", version = "3.8")
library(TxDb.Hsapiens.UCSC.hg19.knownGene)
> TxDb.Hsapiens.UCSC.hg19.knownGene
TxDb object:
# Db type: TxDb
# Supporting package: GenomicFeatures
# Data source: UCSC
# Genome: hg19
# Organism: Homo sapiens
# Taxonomy ID: 9606
# UCSC Table: knownGene
# Resource URL: http://genome.ucsc.edu/
# Type of Gene ID: Entrez Gene ID
# Full dataset: yes
# miRBase build ID: GRCh37
# transcript_nrow: 82960
# exon_nrow: 289969
# cds_nrow: 237533
# Db created by: GenomicFeatures package from Bioconductor
# Creation time: 2015-10-07 18:11:28 +0000 (Wed, 07 Oct 2015)
# GenomicFeatures version at creation time: 1.21.30
# RSQLite version at creation time: 1.0.0
# DBSCHEMAVERSION: 1.1
genes.data <- makeGenesDataFromTxDb(TxDb.Hsapiens.UCSC.hg19.knownGene,
karyoplot=kp,
plot.transcripts = TRUE,
plot.transcripts.structure = TRUE)
kp <- plotKaryotype(zoom = TP53.region) # 第一个图层 染色体绘制的区间
kpPlotGenes(kp, data=genes.data) # 第二个图层 区间内各个基因的结构
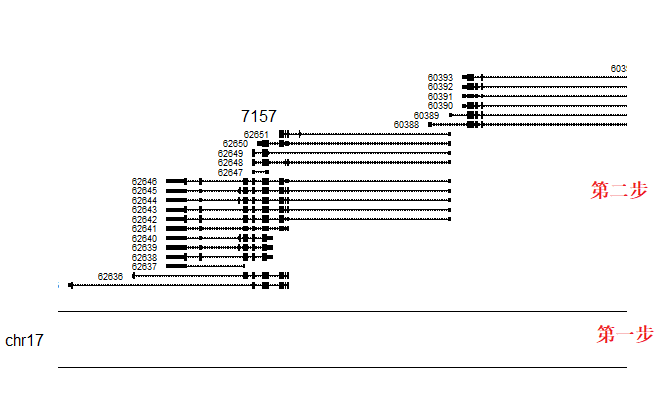
- 从上图可以看到在这个区域内的不同转录本的结构图,而不是基因的结构图,接下来将使用
mergeTranscripts函数将每个基因对应的所有转录本合并成一个,并且使用addGeneNames加上基因名。使用cex函数增加染色体的名字大小。
genes.data <- addGeneNames(genes.data)
genes.data <- mergeTranscripts(genes.data)
> genes.data
$`genes`
GRanges object with 2 ranges and 2 metadata columns:
seqnames ranges strand | gene_id name
|
55135 chr17 7589389-7606820 + | 55135 WRAP53
7157 chr17 7565097-7590868 - | 7157 TP53
-------
seqinfo: 93 sequences (1 circular) from hg19 genome
$transcripts
$transcripts$`55135`
GRanges object with 1 range and 1 metadata column:
seqnames ranges strand | tx_id
|
[1] chr17 7589389-7606820 + | 55135.merged
-------
seqinfo: 93 sequences (1 circular) from hg19 genome
kp <- plotKaryotype(zoom = TP53.region, cex=2)
kpPlotGenes(kp, data=genes.data)
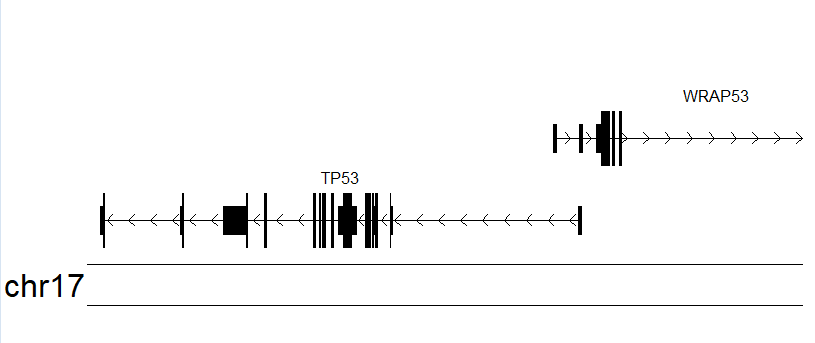
- 这才是对于我们展示基因结构的比较好的方式啊。
- 接下来将使用
r0和r1将基因结构图放置在最下面,为上面其他数据元素留出空间。
kp <- plotKaryotype(zoom = TP53.region, cex=2)
kpPlotGenes(kp, data=genes.data, r0=0, r1=0.15, gene.name.cex = 2)
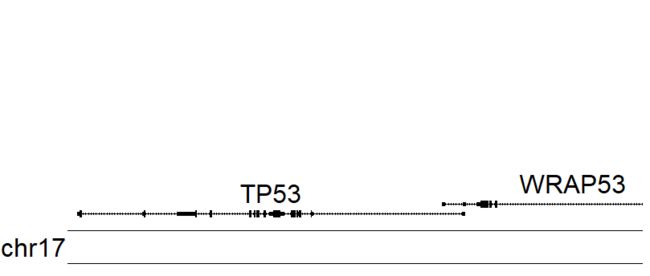
- 下一步将将基因状态HMM结果导入进来,首先使用BiocFileCache 函数下载下来。然后再使用
regioneR包的函数toGranges导入R。
BiocManager::install("BiocFileCache", version = "3.8")
library(BiocFileCache)
bfc <- BiocFileCache(ask=FALSE)
> bfc
class: BiocFileCache
bfccache: C:\Users\ql\AppData\Local\BiocFileCache\BiocFileCache\Cache
bfccount: 0
For more information see: bfcinfo() or bfcquery()
K562.hmm.file <- bfcrpath(bfc, "http://hgdownload.soe.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeBroadHmm/wgEncodeBroadHmmK562HMM.bed.gz")
K562.hmm <- toGRanges(K562.hmm.file)
K562.hmm
> K562.hmm
GRanges object with 622257 ranges and 4 metadata columns:
seqnames ranges strand | name score itemRgb thick
|
[1] chr1 10001-10600 * | 15_Repetitive/CNV 0 #F5F5F5 10001-10600
[2] chr1 10601-10937 * | 13_Heterochrom/lo 0 #F5F5F5 10601-10937
[3] chr1 10938-11937 * | 8_Insulator 0 #0ABEFE 10938-11937
[4] chr1 11938-12337 * | 5_Strong_Enhancer 0 #FACA00 11938-12337
[5] chr1 12338-13137 * | 7_Weak_Enhancer 0 #FFFC04 12338-13137
... ... ... ... . ... ... ... ...
[622253] chrX 155256807-155257806 * | 11_Weak_Txn 0 #99FF66 155256807-155257806
[622254] chrX 155257807-155259206 * | 8_Insulator 0 #0ABEFE 155257807-155259206
[622255] chrX 155259207-155259406 * | 6_Weak_Enhancer 0 #FFFC04 155259207-155259406
[622256] chrX 155259407-155259606 * | 7_Weak_Enhancer 0 #FFFC04 155259407-155259606
[622257] chrX 155259607-155260406 * | 15_Repetitive/CNV 0 #F5F5F5 155259607-155260406
-------
seqinfo: 23 sequences from an unspecified genome; no seqlengths
- 可以从上面看到在
itemRgb列有对应的颜色信息,当我们使用kpPlotRegions函数绘图时候就可以使用这里的颜色来代表区域的颜色。
kp <- plotKaryotype(zoom = TP53.region, cex=2)
kpPlotGenes(kp, data=genes.data, r0=0, r1=0.15, gene.name.cex = 2)
kpPlotRegions(kp, K562.hmm, col=K562.hmm$itemRgb, r0=0.4, r1=0.5)
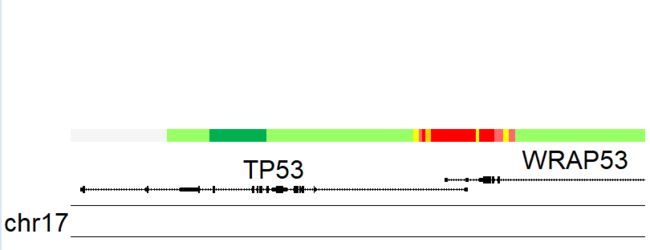
- 使用
kpAddLabels添加HMM注释信息。
kp <- plotKaryotype(zoom = TP53.region, cex=2)
kpPlotGenes(kp, data=genes.data, r0=0, r1=0.15, gene.name.cex = 2)
kpPlotRegions(kp, K562.hmm, col=K562.hmm$itemRgb, r0=0.4, r1=0.5)
kpAddLabels(kp, labels = "Chromatin\nState (HMM)", r0=0.4, r1=0.5, cex = 1)
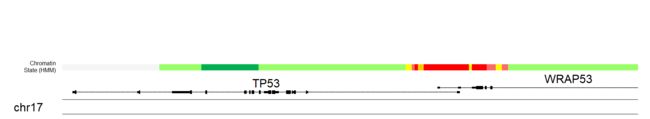
注意控制字体大小
- 接下来我们可以添加一些表观数据了。使用
kpPlotBigwig函数绘制包含BigWig文件的图片。此函数调用的是rtracklayer’s BigWigFile函数来导入BigWig文件。
注意!由于rtracklayer bigwig管理的限制,kpPlotBigWig在Windows上不起作用。它仅适用于Linux和Mac计算机 - 首先我们将可视化H3K4me3信息。绘制在
0.
kp <- plotKaryotype(zoom = TP53.region, cex=2)
kpPlotGenes(kp, data=genes.data, r0=0, r1=0.15, gene.name.cex = 2)
kpPlotRegions(kp, K562.hmm, col=K562.hmm$itemRgb, r0=0.4, r1=0.5)
kpAddLabels(kp, labels = "Chromatin\nState (HMM)", r0=0.4, r1=0.5, cex = 1)
bigwig.file <- "http://hgdownload.soe.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeBroadHistone/wgEncodeBroadHistoneK562H3k4me3StdSig.bigWig"
kpPlotBigWig(kp, data=bigwig.file, r0=0.55, r1=1)
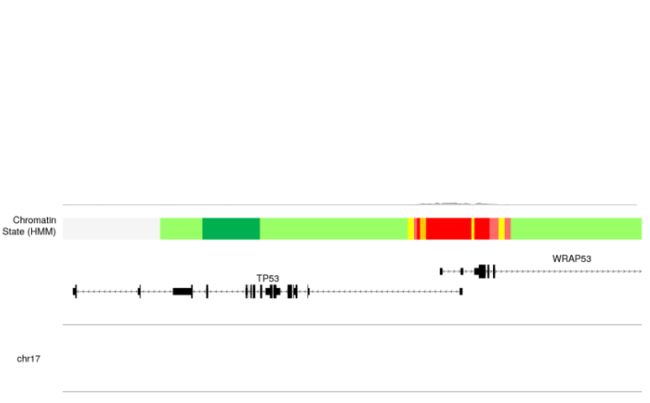
- 可以看到上面bw文件展示出来的信息峰值太低了,因为我们选取的是默认的
ymax, 所以会将y轴的最大值默认为全局的最大值。我们可以通过ymax = "visible.region"来设置。
kp <- plotKaryotype(zoom = TP53.region, cex=2)
kpPlotGenes(kp, data=genes.data, r0=0, r1=0.15, gene.name.cex = 2)
kpPlotRegions(kp, K562.hmm, col=K562.hmm$itemRgb, r0=0.4, r1=0.5)
kpAddLabels(kp, labels = "Chromatin\nState (HMM)", r0=0.4, r1=0.5, cex = 1)
bigwig.file <- "http://hgdownload.soe.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeBroadHistone/wgEncodeBroadHistoneK562H3k4me3StdSig.bigWig"
kpPlotBigWig(kp, data=bigwig.file, ymax="visible.region", r0=0.55, r1=1)
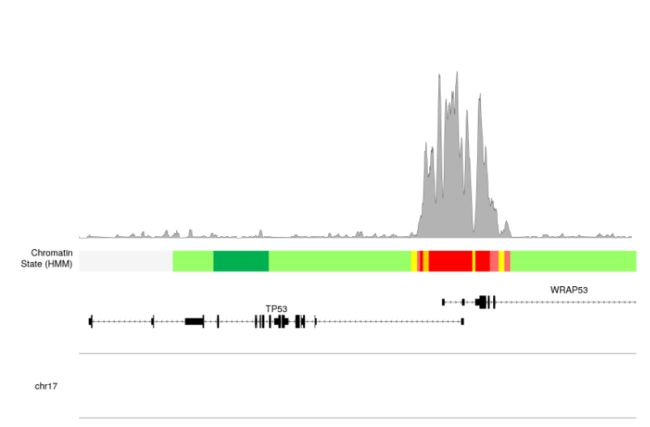
- 接下来我们加入
H3K36me3的信息
kp <- plotKaryotype(zoom = TP53.region, cex=2)
kpPlotGenes(kp, data=genes.data, r0=0, r1=0.15, gene.name.cex = 2)
kpPlotRegions(kp, K562.hmm, col=K562.hmm$itemRgb, r0=0.4, r1=0.5)
kpAddLabels(kp, labels = "Chromatin\nState (HMM)", r0=0.4, r1=0.5, cex = 1)
H3K4me3.bw <- "http://hgdownload.soe.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeBroadHistone/wgEncodeBroadHistoneK562H3k4me3StdSig.bigWig"
kpPlotBigWig(kp, data = H3K4me3.bw, ymax="visible.region", r0=0.55, r1=1)
H3K36me3.bw <- "http://hgdownload.soe.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeBroadHistone/wgEncodeBroadHistoneK562H3k36me3StdSig.bigWig"
kpPlotBigWig(kp, data=H3K36me3.bw, ymax="visible.region", r0 = 1, r1 = 1.3)
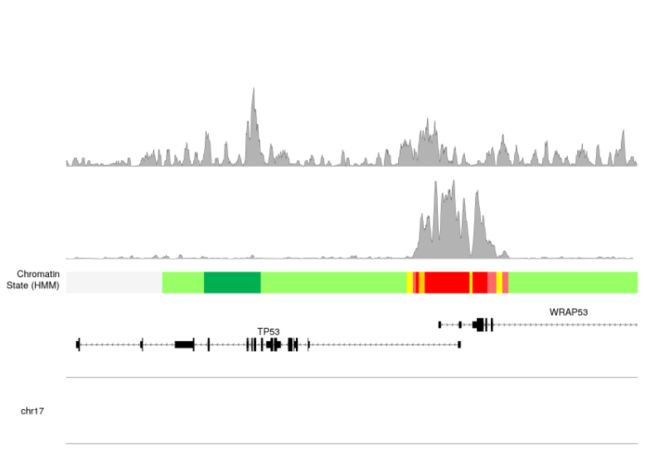
- 我们可以看到不同的peak profiler。但是我们还是缺少一些注释信息,比如每个bw文件对应的修饰信息、峰的高度信息。
kp <- plotKaryotype(zoom = TP53.region, cex=2)
kpPlotGenes(kp, data=genes.data, r0=0, r1=0.15, gene.name.cex = 2)
kpPlotRegions(kp, K562.hmm, col=K562.hmm$itemRgb, r0=0.4, r1=0.5)
kpAddLabels(kp, labels = "Chromatin\nState (HMM)", r0=0.4, r1=0.5, cex = 1)
H3K4me3.bw <- "http://hgdownload.soe.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeBroadHistone/wgEncodeBroadHistoneK562H3k4me3StdSig.bigWig"
kpPlotBigWig(kp, data = H3K4me3.bw, ymax="visible.region", r0=0.55, r1=1)
computed.ymax <- kp$latest.plot$computed.values$ymax
kpAxis(kp, ymin=0, ymax=computed.ymax, r0=0.55, r1=1)
kpAddLabels(kp, labels = "H3K4me3", r0=0.55, r1=1, cex=1.6, label.margin = 0.035)
H3K36me3.bw <- "http://hgdownload.soe.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeBroadHistone/wgEncodeBroadHistoneK562H3k36me3StdSig.bigWig"
kpPlotBigWig(kp, data=H3K36me3.bw, ymax="visible.region", r0 = 1, r1 = 1.3)
computed.ymax <- kp$latest.plot$computed.values$ymax # 获得所绘制区域y的最大值
kpAxis(kp, ymin=0, ymax=computed.ymax, r0 = 1, r1 = 1.3) # 设置Y轴坐标值
kpAddLabels(kp, labels = "H3K36me3", r0 = 1, r1 = 1.3, cex=1.6, label.margin = 0.035) # 设置Y轴标签
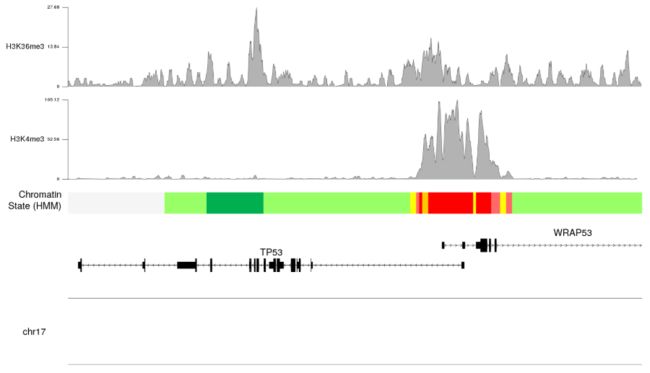
- 我们可以看到
H3K4me3的修饰要高于H3K36me3的修饰的。我们还可以看到,我们已经开始重复代码,并且为此使用循环会更好,我们将使用autotrack函数自动获取r0和r1值。
histone.marks <- c(H3K4me3="wgEncodeBroadHistoneK562H3k4me3StdSig.bigWig",
H3K36me3="wgEncodeBroadHistoneK562H3k36me3StdSig.bigWig")
base.url <- "http://hgdownload.soe.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeBroadHistone/"
kp <- plotKaryotype(zoom = TP53.region, cex=2)
kpPlotGenes(kp, data=genes.data, r0=0, r1=0.15, gene.name.cex = 2)
kpPlotRegions(kp, K562.hmm, col=K562.hmm$itemRgb, r0=0.22, r1=0.3)
kpAddLabels(kp, labels = "Chromatin\nState (HMM)", r0=0.22, r1=0.3, cex=2)
for(i in seq_len(length(histone.marks))) {
bigwig.file <- paste0(base.url, histone.marks[i])
at <- autotrack(i, length(histone.marks), r0=0.35, r1=1)
kp <- kpPlotBigWig(kp, data=bigwig.file, ymax="visible.region",
r0=at$r0, r1=at$r1)
computed.ymax <- ceiling(kp$latest.plot$computed.values$ymax)
kpAxis(kp, ymin=0, ymax=computed.ymax, numticks = 2, r0=at$r0, r1=at$r1)
kpAddLabels(kp, labels = names(histone.marks)[i], r0=at$r0, r1=at$r1,
cex=1.6, label.margin = 0.035)
}
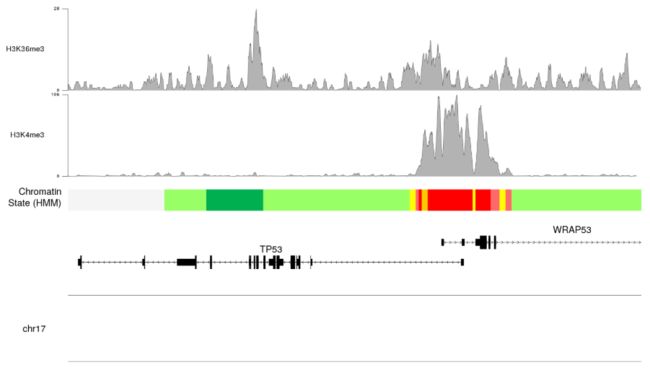
- 一旦我们有for循环和
autotrack自动跟踪到位,我们可以增加组蛋白标记的数量,一切都将自动调整。
histone.marks <- c(H3K4me3="wgEncodeBroadHistoneK562H3k4me3StdSig.bigWig",
H3K36me3="wgEncodeBroadHistoneK562H3k36me3StdSig.bigWig",
H3K27ac="wgEncodeBroadHistoneK562H3k27acStdSig.bigWig",
H3K9ac="wgEncodeBroadHistoneK562H3k9acStdSig.bigWig",
H3K27me3="wgEncodeBroadHistoneK562H3k27me3StdSig.bigWig")
base.url <- "http://hgdownload.soe.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeBroadHistone/"
kp <- plotKaryotype(zoom = TP53.region, cex=2)
kpPlotGenes(kp, data=genes.data, r0=0, r1=0.15, gene.name.cex = 2)
kpPlotRegions(kp, K562.hmm, col=K562.hmm$itemRgb, r0=0.22, r1=0.3)
kpAddLabels(kp, labels = "Chromatin\nState (HMM)", r0=0.22, r1=0.3, cex=2)
for(i in seq_len(length(histone.marks))) {
bigwig.file <- paste0(base.url, histone.marks[i])
at <- autotrack(i, length(histone.marks), r0=0.35, r1=1, margin = 0.1)
kp <- kpPlotBigWig(kp, data=bigwig.file, ymax="visible.region",
r0=at$r0, r1=at$r1)
computed.ymax <- ceiling(kp$latest.plot$computed.values$ymax)
kpAxis(kp, ymin=0, ymax=computed.ymax, numticks = 2, r0=at$r0, r1=at$r1)
kpAddLabels(kp, labels = names(histone.marks)[i], r0=at$r0, r1=at$r1,
cex=1.6, label.margin = 0.035)
}
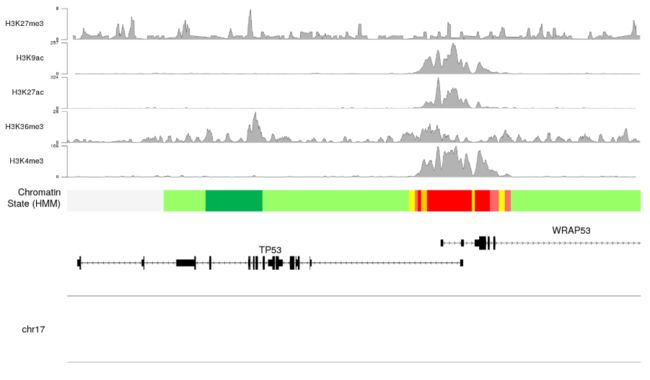
- 我们现在可以调整绘图参数 plotting parameters 来减少边距和表意文字高度并改变颜色以改善plo的整体外观
pp <- getDefaultPlotParams(plot.type=1)
pp$leftmargin <- 0.15
pp$topmargin <- 15
pp$bottommargin <- 15
pp$ideogramheight <- 5
pp$data1inmargin <- 10
kp <- plotKaryotype(zoom = TP53.region, cex=2, plot.params = pp)
kpAddBaseNumbers(kp, tick.dist = 10000, minor.tick.dist = 2000,
add.units = TRUE, cex=1.3, digits = 6)
kpPlotGenes(kp, data=genes.data, r0=0, r1=0.1, gene.name.cex = 2)
kpPlotRegions(kp, K562.hmm, col=K562.hmm$itemRgb, r0=0.15, r1=0.18)
kpAddLabels(kp, labels = "Chromatin\nState (HMM)", r0=0.15, r1=0.18, cex=2)
for(i in seq_len(length(histone.marks))) {
bigwig.file <- paste0(base.url, histone.marks[i])
at <- autotrack(i, length(histone.marks), r0=0.23, r1=1, margin = 0.1)
kp <- kpPlotBigWig(kp, data=bigwig.file, ymax="visible.region",
r0=at$r0, r1=at$r1, col = "cadetblue2")
computed.ymax <- ceiling(kp$latest.plot$computed.values$ymax)
kpAxis(kp, ymin=0, ymax=computed.ymax, numticks = 2, r0=at$r0, r1=at$r1)
kpAddLabels(kp, labels = names(histone.marks)[i], r0=at$r0, r1=at$r1,
cex=1.6, label.margin = 0.035)
}
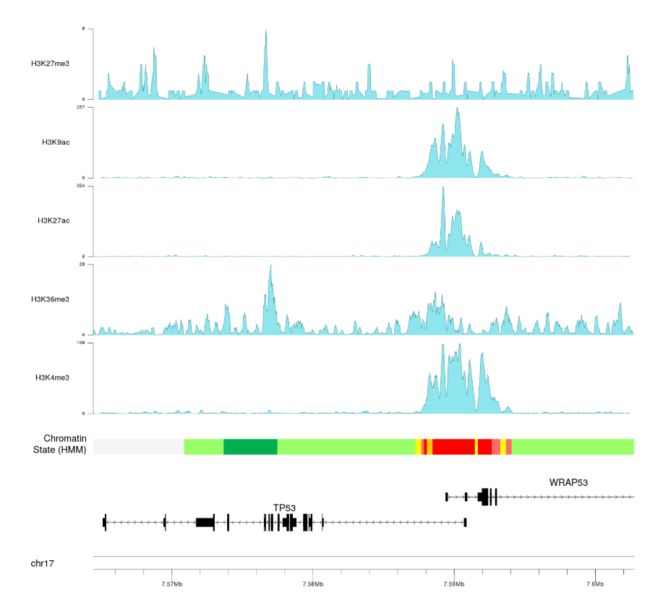
- 我们甚至可以添加其他实验峰值并使用嵌套自动跟踪
autotrack来定位它们
base.url <- "http://hgdownload.soe.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeBroadHistone/"
histone.marks <- c(H3K4me3="wgEncodeBroadHistoneK562H3k4me3StdSig.bigWig",
H3K36me3="wgEncodeBroadHistoneK562H3k36me3StdSig.bigWig",
H3K27ac="wgEncodeBroadHistoneK562H3k27acStdSig.bigWig",
H3K9ac="wgEncodeBroadHistoneK562H3k9acStdSig.bigWig",
H3K27me3="wgEncodeBroadHistoneK562H3k27me3StdSig.bigWig")
DNA.binding <- c(CTCF="wgEncodeBroadHistoneK562CtcfStdSig.bigWig",
EZH2="wgEncodeBroadHistoneK562Ezh239875StdSig.bigWig",
POL2="wgEncodeBroadHistoneK562Pol2bStdSig.bigWig",
P300="wgEncodeBroadHistoneK562P300StdSig.bigWig",
HDAC1="wgEncodeBroadHistoneK562Hdac1sc6298StdSig.bigWig",
HDAC2="wgEncodeBroadHistoneK562Hdac2a300705aStdSig.bigWig")
pp <- getDefaultPlotParams(plot.type=1)
pp$leftmargin <- 0.15
pp$topmargin <- 15
pp$bottommargin <- 15
pp$ideogramheight <- 5
pp$data1inmargin <- 10
kp <- plotKaryotype(zoom = TP53.region, cex=2, plot.params = pp)
kpAddBaseNumbers(kp, tick.dist = 10000, minor.tick.dist = 2000,
add.units = TRUE, cex=1.3, digits = 6)
kpPlotGenes(kp, data=genes.data, r0=0, r1=0.1, gene.name.cex = 2)
kpPlotRegions(kp, K562.hmm, col=K562.hmm$itemRgb, r0=0.15, r1=0.18)
kpAddLabels(kp, labels = "Chromatin\nState (HMM)", r0=0.15, r1=0.18, cex=2)
#Histone marks
total.tracks <- length(histone.marks)+length(DNA.binding)
out.at <- autotrack(1:length(histone.marks), total.tracks, margin = 0.3, r0=0.23)
for(i in seq_len(length(histone.marks))) {
bigwig.file <- paste0(base.url, histone.marks[i])
at <- autotrack(i, length(histone.marks), r0=out.at$r0, r1=out.at$r1, margin = 0.1)
kp <- kpPlotBigWig(kp, data=bigwig.file, ymax="visible.region",
r0=at$r0, r1=at$r1, col = "cadetblue2")
computed.ymax <- ceiling(kp$latest.plot$computed.values$ymax)
kpAxis(kp, ymin=0, ymax=computed.ymax, numticks = 2, r0=at$r0, r1=at$r1)
kpAddLabels(kp, labels = names(histone.marks)[i], r0=at$r0, r1=at$r1,
cex=1.6, label.margin = 0.035)
}
#DNA binding proteins
out.at <- autotrack((length(histone.marks)+1):total.tracks, total.tracks, margin = 0.3, r0=0.23)
for(i in seq_len(length(DNA.binding))) {
bigwig.file <- paste0(base.url, DNA.binding[i])
at <- autotrack(i, length(DNA.binding), r0=out.at$r0, r1=out.at$r1, margin = 0.1)
kp <- kpPlotBigWig(kp, data=bigwig.file, ymax="visible.region",
r0=at$r0, r1=at$r1, col = "darkolivegreen1")
computed.ymax <- ceiling(kp$latest.plot$computed.values$ymax)
kpAxis(kp, ymin=0, ymax=computed.ymax, numticks = 2, r0=at$r0, r1=at$r1)
kpAddLabels(kp, labels = names(DNA.binding)[i], r0=at$r0, r1=at$r1,
cex=1.6, label.margin = 0.035)
}
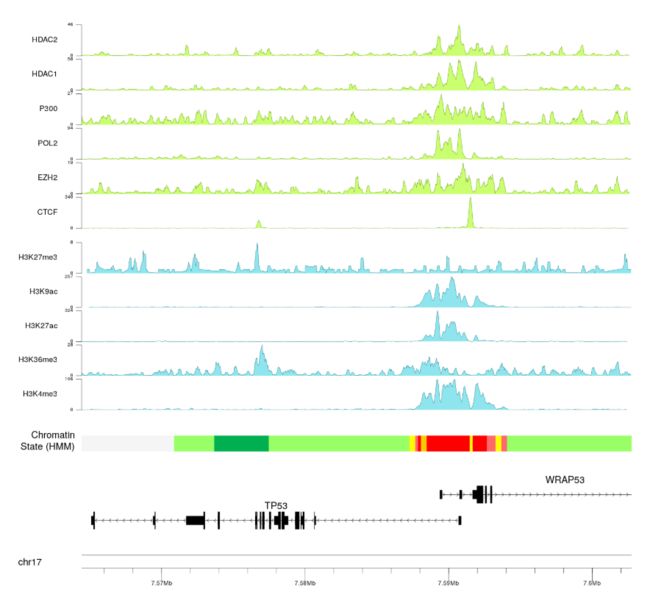
- 并添加一个主标题,几个额外的标签,并调整一些参数(文字大小等...),以获得更好的最终图像
base.url <- "http://hgdownload.soe.ucsc.edu/goldenPath/hg19/encodeDCC/wgEncodeBroadHistone/"
histone.marks <- c(H3K4me3="wgEncodeBroadHistoneK562H3k4me3StdSig.bigWig",
H3K36me3="wgEncodeBroadHistoneK562H3k36me3StdSig.bigWig",
H3K27ac="wgEncodeBroadHistoneK562H3k27acStdSig.bigWig",
H3K9ac="wgEncodeBroadHistoneK562H3k9acStdSig.bigWig",
H3K27me3="wgEncodeBroadHistoneK562H3k27me3StdSig.bigWig")
DNA.binding <- c(CTCF="wgEncodeBroadHistoneK562CtcfStdSig.bigWig",
EZH2="wgEncodeBroadHistoneK562Ezh239875StdSig.bigWig",
POL2="wgEncodeBroadHistoneK562Pol2bStdSig.bigWig",
P300="wgEncodeBroadHistoneK562P300StdSig.bigWig",
HDAC1="wgEncodeBroadHistoneK562Hdac1sc6298StdSig.bigWig",
HDAC2="wgEncodeBroadHistoneK562Hdac2a300705aStdSig.bigWig")
pp <- getDefaultPlotParams(plot.type=1)
pp$leftmargin <- 0.15
pp$topmargin <- 15
pp$bottommargin <- 15
pp$ideogramheight <- 5
pp$data1inmargin <- 10
pp$data1outmargin <- 0
kp <- plotKaryotype(zoom = TP53.region, cex=3, plot.params = pp)
kpAddBaseNumbers(kp, tick.dist = 10000, minor.tick.dist = 2000,
add.units = TRUE, cex=2, tick.len = 3)
kpAddMainTitle(kp, "Epigenetic Regulation in K562", cex=4)
kpPlotGenes(kp, data=genes.data, r0=0, r1=0.1, gene.name.cex = 2.5)
kpPlotRegions(kp, K562.hmm, col=K562.hmm$itemRgb, r0=0.15, r1=0.18)
kpAddLabels(kp, labels = "Chromatin\nState (HMM)", r0=0.15, r1=0.18, cex=2.5)
#Histone marks
total.tracks <- length(histone.marks)+length(DNA.binding)
out.at <- autotrack(1:length(histone.marks), total.tracks, margin = 0.3, r0=0.23)
kpAddLabels(kp, labels = "Histone marks", r0 = out.at$r0, r1=out.at$r1, cex=3.5,
srt=90, pos=1, label.margin = 0.14)
for(i in seq_len(length(histone.marks))) {
bigwig.file <- paste0(base.url, histone.marks[i])
at <- autotrack(i, length(histone.marks), r0=out.at$r0, r1=out.at$r1, margin = 0.1)
kp <- kpPlotBigWig(kp, data=bigwig.file, ymax="visible.region",
r0=at$r0, r1=at$r1, col = "cadetblue2")
computed.ymax <- ceiling(kp$latest.plot$computed.values$ymax)
kpAxis(kp, ymin=0, ymax=computed.ymax, tick.pos = computed.ymax,
r0=at$r0, r1=at$r1, cex=1.6)
kpAddLabels(kp, labels = names(histone.marks)[i], r0=at$r0, r1=at$r1,
cex=2.2, label.margin = 0.035)
}
#DNA binding proteins
out.at <- autotrack((length(histone.marks)+1):total.tracks, total.tracks, margin = 0.3, r0=0.23)
kpAddLabels(kp, labels = "DNA-binding proteins", r0 = out.at$r0, r1=out.at$r1,
cex=3.5, srt=90, pos=1, label.margin = 0.14)
for(i in seq_len(length(DNA.binding))) {
bigwig.file <- paste0(base.url, DNA.binding[i])
at <- autotrack(i, length(DNA.binding), r0=out.at$r0, r1=out.at$r1, margin = 0.1)
kp <- kpPlotBigWig(kp, data=bigwig.file, ymax="visible.region",
r0=at$r0, r1=at$r1, col = "darkolivegreen1")
computed.ymax <- ceiling(kp$latest.plot$computed.values$ymax)
kpAxis(kp, ymin=0, ymax=computed.ymax, tick.pos = computed.ymax,
r0=at$r0, r1=at$r1, cex=1.6)
kpAddLabels(kp, labels = names(DNA.binding)[i], r0=at$r0, r1=at$r1,
cex=2.2, label.margin = 0.035)
}
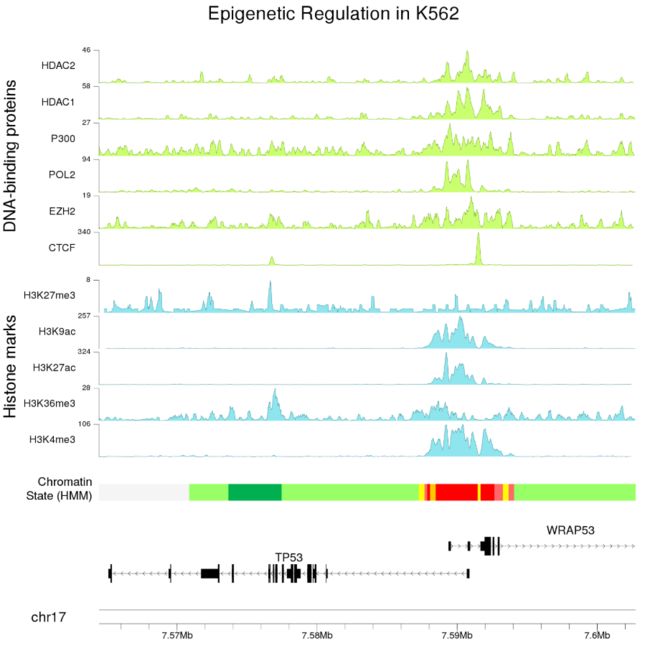
- 与
karyoploteR一样,我们可以更改绘图区域(在这种情况下放大)以绘制基因组的任何部分,例如,重叠区域的详细视图。
TP53.promoter.region <- toGRanges("chr17:7586000-7596000")
kp <- plotKaryotype(zoom = TP53.promoter.region, cex=3, plot.params = pp)
kpAddBaseNumbers(kp, tick.dist = 10000, minor.tick.dist = 2000,
add.units = TRUE, cex=2, tick.len = 3)
kpAddMainTitle(kp, "Epigenetic Regulation in K562", cex=4)
kpPlotGenes(kp, data=genes.data, r0=0, r1=0.1, gene.name.cex = 2.5)
kpPlotRegions(kp, K562.hmm, col=K562.hmm$itemRgb, r0=0.15, r1=0.18)
kpAddLabels(kp, labels = "Chromatin\nState (HMM)", r0=0.15, r1=0.18, cex=2.5)
#Histone marks
total.tracks <- length(histone.marks)+length(DNA.binding)
out.at <- autotrack(1:length(histone.marks), total.tracks, margin = 0.3, r0=0.23)
kpAddLabels(kp, labels = "Histone marks", r0 = out.at$r0, r1=out.at$r1, cex=3.5,
srt=90, pos=1, label.margin = 0.14)
for(i in seq_len(length(histone.marks))) {
bigwig.file <- paste0(base.url, histone.marks[i])
at <- autotrack(i, length(histone.marks), r0=out.at$r0, r1=out.at$r1, margin = 0.1)
kp <- kpPlotBigWig(kp, data=bigwig.file, ymax="visible.region",
r0=at$r0, r1=at$r1, col = "cadetblue2")
computed.ymax <- ceiling(kp$latest.plot$computed.values$ymax)
kpAxis(kp, ymin=0, ymax=computed.ymax, tick.pos = computed.ymax,
r0=at$r0, r1=at$r1, cex=1.6)
kpAddLabels(kp, labels = names(histone.marks)[i], r0=at$r0, r1=at$r1,
cex=2.2, label.margin = 0.035)
}
#DNA binding proteins
out.at <- autotrack((length(histone.marks)+1):total.tracks, total.tracks, margin = 0.3, r0=0.23)
kpAddLabels(kp, labels = "DNA-binding proteins", r0 = out.at$r0, r1=out.at$r1,
cex=3.5, srt=90, pos=1, label.margin = 0.14)
for(i in seq_len(length(DNA.binding))) {
bigwig.file <- paste0(base.url, DNA.binding[i])
at <- autotrack(i, length(DNA.binding), r0=out.at$r0, r1=out.at$r1, margin = 0.1)
kp <- kpPlotBigWig(kp, data=bigwig.file, ymax="visible.region",
r0=at$r0, r1=at$r1, col = "darkolivegreen1")
computed.ymax <- ceiling(kp$latest.plot$computed.values$ymax)
kpAxis(kp, ymin=0, ymax=computed.ymax, tick.pos = computed.ymax,
r0=at$r0, r1=at$r1, cex=1.6)
kpAddLabels(kp, labels = names(DNA.binding)[i], r0=at$r0, r1=at$r1,
cex=2.2, label.margin = 0.035)
}