FACTERA (Fusion And Chromosomal Translocation Enumeration and Recovery Algorithm) is a tool for detection of genomic fusions in paired-end targeted (or genome-wide) sequencing data.
一、概述
FACTERA (PERL编写)
官网或下载地址:
https://factera.stanford.edu/
发表的文章、影响因子及被引用量:
Newman A M , Bratman S V , Stehr H , et al. FACTERA: a practical method for the discovery of genomic rearrangements at breakpoint resolution[J]. Bioinformatics, 2014, 30(23):3390-3393.
发表时间:2014
被引量:14
期刊及影响因子:Bioinformatics 4.531(2018)
作者及单位:Newman A M ;Stanford University Contact: [email protected] or diehn@stanford.
二、下载及安装
官网可下载:https://factera.stanford.edu/download.php#download
目前最新版本:1.4.4
下载得到factera-v1.4.4.zip
解压后,factera.pl即可使用
依赖软件官网介绍的很详细,需要写在环境中,也可以修改源代码~
主要就是perl(需要包含Statistics::Descriptive模块);blast+(用到blastn和makeblastdb);SAMtools;twoBitToFa
Unix operating system (Linux, Mac OS X, etc.)
Perl 5, with the following external dependency: Statistics::Descriptive.
To install Statistics::Descriptive from CPAN, issue the following command:
sudo cpan Statistics::Descriptive
Other Perl dependencies are included in the Perl 5 Core Modules and should already be installed: IPC::Open3, List::Util, File::Spec, Symbol, Getopt::Std, File::Basename.
twoBitToFa
Find and download executable from the appropriate system folder, then copy/link/move to PATH (i.e., /usr/bin).
hg19.2bit to run FACTERA on the hg19 human genome.
Note that hg38.2bit is now available. To use another reference genome, make sure that input BED coordinates are consistent (the exons.bed file provided here is currently hg19 only).
blast+
After downloading, find blastn and makeblastdb in ncbi-blast-version/bin and copy/link/move to PATH (i.e., /usr/bin).
SAMtools
After downloading, find samtools and copy/link/move to PATH (i.e., /usr/bin).
需要注意,FACTERA软件推荐使用bwa aln算法,而不推荐mem
官网解释:
FACTERA was developed and optimized using targeted sequencing data aligned by bwa aln, and we currently recommend that users employ bwa aln for best performance. While FACTERA can be applied to data mapped by bwa mem, users should be aware of the following considerations when interpreting results. The most notable difference between bwa aln and mem with respect to fusion detection is the use of hard clipping in addition to soft clipping by bwa mem. Absent from bwa aln, hard clipping enables bwa mem to improve the mapping rate by realigning (rather than truncating) sufficiently long read segments in chimeric sequences. In contrast, bwa aln will truncate such reads without realignment (soft-clipping), and FACTERA leverages soft clipped, but not hard clipped, reads for breakpoint detection. Hard clipped reads will be supported in a future release of FACTERA, and we will notify registered users when this version is available
三、软件参数介绍
perl factera.pl [options] tumor.bam exons.bed hg19.2bit [optional: targets.bed]
需要注意的是,[options]需要放在factera.pl之后,而不能放在tumor.bam exons.bed hg19.2bit后面。否则参数导入失败。
一)所有参数
FACTERA version 1.4.4 by Aaron M. Newman ([email protected])
Purpose:
A tool for detection of fusions in paired-end targeted (or genome-wide) sequencing data.
Usage:
factera.pl [options]
Paired reads mapped with BWA or another soft-clip capable aligner.
NOTE: Must be indexed for factera to function properly.
Genomic coordinates with gene/exon names in fourth column.
Data sources include RefSeq, UCSC, and/or GENCODE.
Two bit reference sequence (e.g., hg19.2bit).
NOTE: twoBitToFa application must be in PATH.
Restrict search to specified genome coordinates.
Options (defaults in parentheses):
-o output directory (tumor.bam directory) ###输出目录
-r minimum number of breakpoint-spanning reads required for output (5) ###断点最小支持数
-m minimum number of discordant reads required for each candidate fusion (2)
-x maximum number of breakpoints to examine for each pair of genomic regions (5)
-s minimum number of reads with the same breakpoint (1)
-f <0-1> minimum fraction of read bases required for alignment to fusion template (0.9)
-S <0-1> minimum similarity required for read to match fusion template (0.95)
-k k-mer size for fragment comparison (12 bases)
-c minimum size of soft-clipped region to consider (16 bases)
-b number of bases flanking breakpoint for fusion template (500)
-p number of threads for blastn search (4; 10 or more recommended)
-a number of bases flanking breakpoint to provide in output (50)
-e disable grouping of input coordinates by column 4 of exons.bed (off)
-v disable verbose output (off)
-t disable running time output (off)
-C disable addition of 'chr' prefix to chromosome names (off)
-F force remake of BLAST database for a particular input (off)
For detailed help, see http://factera.stanford.edu
二)几个关键参数的解释:
1)
should consist of paired-end reads aligned by a mapping algorithm capable of soft-clipping, such as BWA. The BAM file does not need to be realigned or deduped, but should be position-sorted and have a corresponding index file (bam.bai created using SAMtools index) in the same directory in order to estimate the total sequencing depth in the neighborhood of each detected fusion
软件建议用BWA得到的比对结果,因为包含了PE和SR信息。
bam需要进行sort和Index,不无需rmdup和realigned。这样得到的总深度更准确。
2)
contains chromosomal coordinates (such as exon boundaries) in 3-column BED format (chr start end). The fourth column contains gene names, exon names, or any arbitrary identifier, and will be used to group corresponding coordinates. This allows the resolution of fusion detection to be restricted to inter-gene or inter-exon fusions, for example. Make sure to use coordinates from the same genome version as the 2bit reference sequence required in the third argument.
需要包含至少三列信息,染色体,exon起始位置。
第四列是其他信息,可以用于区域分组。可以用-e选项禁用这一功能。
给定这个文件后,得到的融合结果是再这些gene和exon之前的区域,需要注意是,该文件位置需要和参考基因组bit文件版本一致。(理清hg19/hg38版本)
3)
hg19.2bit is a 2 bit encoded human reference genome, used for fast genome subsequence retrieval. Of note, FACTERA is not restricted to human sequences, and any 2bit reference genome can be used as long as coordinates in exons.bed are consistent. To create a 2bit file for a genome of interest, download the FASTA to 2BIT conversion tool from the appropriate system folder (faToTwoBit) and follow these instructions.
从fasta到2bit的转换如下:
wget http://hgdownload.cse.ucsc.edu/admin/exe/linux.x86_64/faToTwoBit
chmod a+x faToTwoBit
/data/Project/root/tools/FACTERA/faToTwoBit ucsc.hg19.fasta ucsc.hg19.fasta.2bit
4)
targets.bed is optional and allows the user to restrict the FACTERA search to genomic regions of interest, such as those targeted by a sequencing capture library. Format is a standard 3-column BED (chr start end). The use of a targets.bed file can greatly improve running time when only a subset of sequenced regions is known to be relevant for fusion detection.
可选项。可以是捕获探针的区域。包含Bed的三列,染色体和起始位置。
四、用法及结果说明
一)用法示例
perl factera.pl -F -r 2 -m 1 -x 200 -f 0.8 -S 0.8 -o FACTERA samplename.rmdup.sort.bam /data/Project/cailili/Gene27/bin/software/FACTERA/exons.bed ucsc.hg19.fasta.2bit panel_27.bed
得到:
-rw-rw-r--. 1 root root 1047 Sep 17 09:22 samplename.sort.factera.blastquery.fa
-rw-rw-r--. 1 root root 19925450 Sep 17 09:22 samplename.sort.factera.blastreads.fa
-rw-rw-r--. 1 root root 12011885 Sep 17 09:22 samplename.sort.factera.blastreads.fa.nhr
-rw-rw-r--. 1 root root 1213668 Sep 17 09:22 samplename.sort.factera.blastreads.fa.nin
-rw-rw-r--. 1 root root 3578331 Sep 17 09:22 samplename.sort.factera.blastreads.fa.nsq
-rw-rw-r--. 1 root root 26588 Sep 17 09:21 samplename.sort.factera.discordantpair.depth.txt
-rw-rw-r--. 1 root root 2015295 Sep 17 09:21 samplename.sort.factera.discordantpair.details.txt
-rw-rw-r--. 1 root root 380 Sep 17 09:22 samplename.sort.factera.fusions.bed
-rw-rw-r--. 1 root root 3140 Sep 17 09:22 samplename.sort.factera.fusionseqs.fa
-rw-rw-r--. 1 root root 803 Sep 17 09:22 samplename.sort.factera.fusions.txt
-rw-rw-r--. 1 root root 38446 Sep 17 09:21 samplename.sort.factera.fusiontargets.bed
-rw-rw-r--. 1 root root 1200 Sep 17 09:21 samplename.sort.factera.parameters.txt
二)结果解读
最后需要的文件是samplename.sort.factera.fusions.txt 其他都是中间文件
$ cat samplename.sort.factera.fusions.txt
Est_Type Region1 Region2 Break1 Break2 Break_support1 Break_support2 Break_offset Orientation Order1 Order2 Break_depth Proper_pair_support Unmapped_support Improper_pair_support Paired_end_depth Total_depth Fusion_seq Non-templated_seq
INV ALK EML4 chr2:29447601 chr2:42524142 156 23 -48 1- 2+ CN CN 87 15 0 72 0 2493 CCATATGGTGCCATCCCTCAAAGGGACAGGATAATAGGAGCTAACACTTG <> TTGAATTAGACCTATTAAGAAGTTTAAAACAAGAAGCCTTAAATTGTATT -
TRA TFAP2B BCL2L11 chr6:50921898 chr2:111886423 1 3 -14 1+ 2- NC NC 55 17 0 38 0 735 AGGCTTCTCTTTCAATCTAGAAATTGAAAGTAAAGTAAGAAAAAGAGAAA <> GTCAATATTAAAATACATTTTATTTAATCACCAATGACCATATGACAAAA -
TRA FGFR1 AL627309.2 chr8:38318668 chr1:16526 2 1 1 1+ 2+ NC CN 12 3 0 9 0 TCCATCACAGGTGGTTATGAAGACTGAATGGGCTGCATGGGTGAAAGTGT <> CCACTTAACAAACCCACAGAAAATCCACCCGAGTGCACTGAGCACGCCAG [T]
需要关注的是有以下几列:
Est_Type,变异类型,该软件有三种:TRA INV DEL (translocations, inversions and deletions)
Region1,基因1
Region2,基因2
Break1,基因1断点位置
Break2,基因2断点位置
Break_support1,基因1断点支持reads
Break_support2,基因2断点支持reads
Break_depth,断点深度
Proper_pair_support,reads两端都匹配的深度
Improper_pair_support,非reads两端都匹配的深度 和Proper_pair_support加起来是Break_depth
Paired_end_depth,read1 read2同时支持该断点的深度
Total_depth 总深度
五、文献解读
FACTERA: a practical method for the discovery of genomic rearrangements at breakpoint resolution
一)引言
Here, we describe and benchmark FACTERA, a new software tool for the discovery of genomic rearrangements, including translocations, inversions and deletions.
之前检测融合基因的软件运行良好,但是往往高估断点支持数。FACTERA在特异性不降低的基础上,使得检测灵敏度性增加。
二)方法
软件检测的原理图如下所示:
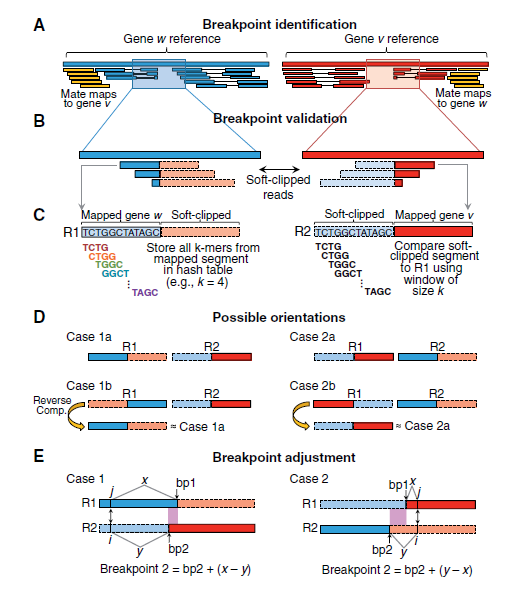
As input, FACTERA requires (i) a Binary Alignment/Map (BAM) file of pairedend reads mapped by an alignment tool capable of ‘soft clipping’, such as Burrows-Wheeler Aligner (BWA) (Li and Durbin, 2009), (ii) genomic coordinates (in Browser Extensible Data [BED] format) used to control the resolution of fusion discovery via the locations of genes, exons or other genomic units and (iii) a 2BIT reference genome to enable fast sequence retrieval (e.g. UCSC hg19.2 bit).
FACTERA需要三个必须文件,1)bam文件,包含PE比对的soft clipping情况;2)bed文件,能够精准定位基因位置;3)bit2文件,能够快速读取基因组文件。
Input BAM files are processed in three key phases: identification of discordant read clusters, detection of breakpoints at nucleotide resolution and in silico validation of candidate fusions.
输入的bam需要经过三个关键步骤:1)鉴定discordant read ;2)鉴定断点在基因组上的位置 ;3)候选融合的确定
工作原理总共分为A-E五步:
A:
确定Ri(潜在的融合基因区域)
确定方法:For every group, a genomic region Ri is defined for each gene by taking the minimum of all 30 coordinates in the cluster (exons and discordant reads) and the maximum of all 50 coordinates in the same.
B:
寻找‘soft-clipped’reads,用来确定断点位置。这种reads支持数默认是5,可以在参数中修改。
C:
对于断点的组合,会筛选两种情况作为候选断点1)断点接近于整条raeds的一半;2)R2的软比对端长度>=15bp,否则认为是非特异性比对
如果该位置是一个断点,那么R1能够比对到R2的soft-clipped部分,反之亦然。
采用滑动窗口(k=10)的形式,对R1序列打断成kmer(kmer=4),将带有index的kmer 储存在哈希中。用这些kmer比对R2的soft-clipped区域,如果比对符合最小阈值,则认为该位置是一个融合候选位点。
D:
列举了四种R1/R2的方向情况,只有两种情况可以被检测到融合,所以需要再C之前,将reads方向转化为所需方向。
F:
由于断点周围的短序列存在同源或者相似序列,会妨碍融合的鉴定。在融合断点鉴定的时候,有允许的偏移量。
最后,会用blastn进行reads和候选融合序列的比对。需要达到95%相似性及90%match覆盖度。
此外,还有一些细节的算法优化,如下:
In addition to the basic algorithm, several heuristics were implemented to improve performance. First, to increase specificity, k-mer comparison is used to assess similarity between the soft-clipped portion of R1 and mapped portion of R2 in addition to the opposite scenario shown in Figure 1C. The same matching threshold described above is required for further consideration of a candidate fusion. Moreover, if breakpoint adjustment is applied initially (Fig. 1E), an equal but opposite breakpoint offset is required for the reciprocal comparison in order for the candidate fusion to proceed. Second, to suppress errors, a consensus sequence is derived from soft-clipped segments that share the same putative breakpoint (e.g. Fig. 1C), and this ‘corrected’ sequence is used for read comparison. Third, if breakpoint adjustment is required for R2, the subsequence in R2 between both original breakpoints (i.e. bp1 and bp2 in Fig. 1E) is compared with the corresponding sequence in the reference genome. If the two sequences are identical, the breakpoint adjustment is performed to R2 (i.e. gene 2). Otherwise, an equal but opposite breakpoint adjustment is performed to R1 (i.e. gene 1), while no adjustment is made for R2. This subroutine reduces the impact of alignment errors on breakpoint adjustment. For further details, including implementation and output, see Supplementary Notes.
三)结果
将FACTERA用于已知的样本检测,效果可观。并和五种软件相比,表现出了高灵敏度和特异性。
用FISH和qPCR等均对FACTERA结果进行了鉴定。
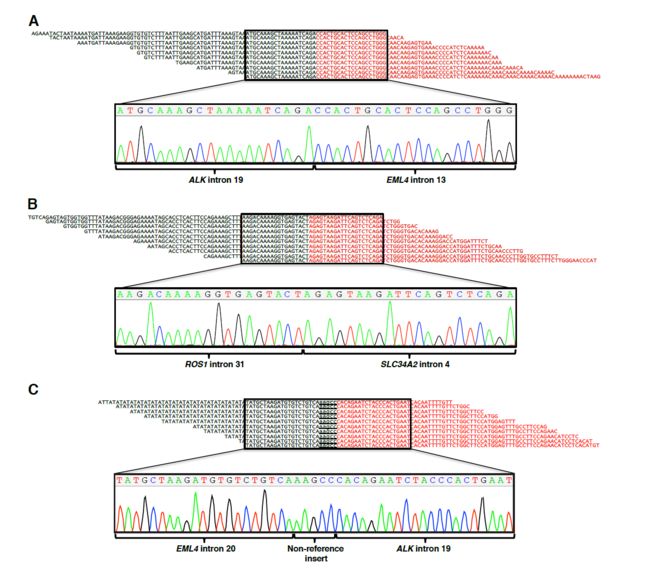
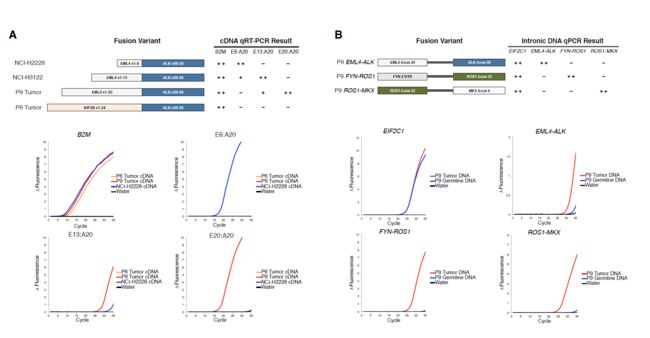
如图: