一文掌握GSEA,超详细教程!
生信宝典之前总结了一篇关于GSEA富集分析的推文——GSEA富集分析:从概念理解到界面实操,介绍了GSEA的定义、GSEA原理、GSEA分析、Leading-edge分析等,是全网最流行的原理+操作兼备教程,不太了解的朋友可以点击阅读先理解下概念 (为了完整性,下面也会摘录一部分)。

GSEA案例解析
介绍GSEA分析之前,我们先看一篇Cell文章(https://sci-hub.tw/10.1016/j.cell.2016.11.033)的一个插图 (SCI-HUB客户端(文献神器V4.0)——下载文献如此简单)。

以下是文章原文对图的注解:GSEA analyses of genesets for cardiac (top) and endothelial/endocardial (bottom) development. NES, normalized enrichment score. FDR, false discovery rate. Positive and negative NES indicate higher and lower expression in iwt, respectively.
关于文章中使用的GSEA分析方法和参数,我们截取对应原文:Gene Set Enrichment Analysis was performed using the GSEA software (https://www.broadinstitute.org/gsea/) with permutation = geneset, metric = Diff_of_classes, metric = weighted, #permutation = 2500.
根据以上信息可知,上图是研究者使用GSEA软件所做的分析结果。文章通过GSEA分析,发现
与心脏发育有关的基因集 (影响心脏的收缩力、钙离子调控和新陈代谢活力等)在
iwt组 (GATA基因野生型)中普遍表达更高,而在G296S组 (GATA基因的一种突变体)中表达更低;而对于参与内皮或内膜发育的基因集,在
iwt组中表达更低,在G296S组中表达更高。作者根据这个图和其它证据推测
iwt组的心脏发育更加完善,而G296S组更倾向于心脏内皮或内膜的发育,即GATA基因的这种突变可能导致心脏内皮或内膜的过度发育而导致心脏相关疾病的产生。
那么GSEA分析是什么?
参考GSEA官网主页的描述:Gene Set Enrichment Analysis (GSEA) is a computational method that determines whether an a priori defined set of genes shows statistically significant, concordant differences between two biological states (e.g. phenotypes).
在上述Cell文章中,作者更加关心参与心脏发育的基因集 (即a priori defined set of genes)与两个状态(突变体和野生型,状态的度量方式是基因表达)的关系,因此利用GSEA对其进行分析后发现,参与心脏发育 (收缩力、钙调控和新陈代谢)的基因集的表达模式更接近于iwt组的表型,而不是G296S组; 而参与心脏内皮或内膜发育的这些基因的表达模式更接近于G296S组的表型而不是iwt组的表型。
这就是GSEA分析所适用的主要场景之一。它能帮助生物学家在两种不同的生物学状态 (biological states)中,判断某一组有特定意义的基因集合的表达模式更接近于其中哪一种。因此GSEA是一种非常常见且实用的分析方法,可以将数个基因组成的基因集与整个转录组、修饰组等做出简单而清晰的关联分析。
除了对特定gene set的分析,反过来GSEA也可以用于发现两组样本从表达或其它度量水平分别与哪些特定生物学意义的基因集有显著关联,或者发现哪些基因集的表达模式或其他模式更接近于表型A、哪些更接近于表型B。这些特定的基因集合可以从GO、KEGG、Reactome、hallmark或MSigDB等基因集中获取,其中MSigDB数据库整合了上述所有基因集。研究者也可自定义gene set (即新发现的基因集或其它感兴趣的基因的集合)。
GSEA分析似乎与GO分析类似但又有所不同。GO分析更加依赖差异基因,实则是对一部分基因的分析 (忽略差异不显著的基因),而GSEA是从全体基因的表达矩阵中找出具有协同差异 (concordant differences)的基因集,故能兼顾差异较小的基因。因此二者的应用场景略有区别。另外GO富集是定性的分析,GSEA考虑到了表达或其它度量水平的值的影响。另外,对于时间序列数据或样品有定量属性时,GSEA的优势会更明显,不需要每个分组分别进行富集,直接对整体进行处理。可以类比于之前的WGCNA分析。
GSEA定义
Gene Set Enrichment Analysis (基因集富集分析)用来评估一个预先定义的基因集的基因在与表型相关度排序的基因表中的分布趋势,从而判断其对表型的贡献。其输入数据包含两部分,一是已知功能的基因集 (可以是GO注释、MsigDB的注释或其它符合格式的基因集定义),一是表达矩阵 (也可以是排序好的列表),软件会对基因根据其与表型的关联度(可以理解为表达值的变化)从大到小排序,然后判断基因集内每条注释下的基因是否富集于表型相关度排序后基因表的上部或下部,从而判断此基因集内基因的协同变化对表型变化的影响。
(The gene sets are defined based on prior biological knowledge, e.g., published information about biochemical pathways or coexpression in previous experiments. The goal of GSEA is to determine whether members of a gene set S tend to occur toward the top (or bottom) of the list L, in which case the gene set is correlated with the phenotypic class distinction.)
这与之前讲述的GO富集分析不同。GO富集分析是先筛选差异基因,再判断差异基因在哪些注释的通路存在富集;这涉及到阈值的设定,存在一定主观性并且只能用于表达变化较大的基因,即我们定义的显著差异基因。而GSEA则不局限于差异基因,从基因集的富集角度出发,理论上更容易囊括细微但协调性的变化对生物通路的影响,尤其是差异倍数不太大的基因集。
GSEA原理
给定一个排序的基因表L和一个预先定义的基因集S (比如编码某个代谢通路的产物的基因, 基因组上物理位置相近的基因,或同一GO注释下的基因),GSEA的目的是判断S里面的成员s在L里面是随机分布还是主要聚集在L的顶部或底部。这些基因排序的依据是其在不同表型状态下的表达差异,若研究的基因集S的成员显著聚集在L的顶部或底部,则说明此基因集成员对表型的差异有贡献,也是我们关注的基因集。

GSEA计算中几个关键概念:
计算富集得分 (ES, enrichment score). ES反应基因集成员
s在排序列表L的两端富集的程度。计算方式是,从基因集L的第一个基因开始,计算一个累计统计值。当遇到一个落在s里面的基因,则增加统计值。遇到一个不在s里面的基因,则降低统计值。每一步统计值增加或减少的幅度与基因的表达变化程度(更严格的是与基因和表型的关联度,可能是fold-change,也可能是pearson corelation值,后面有介绍几种不同的计算方式)是相关的,可以是线性相关,也可以是指数相关 (具体见后面参数选择)。富集得分ES最后定义为最大的峰值。
正值ES表示基因集在列表的顶部富集,负值ES表示基因集在列表的底部富集。评估富集得分(ES)的显著性。通过基于表型而不改变基因之间关系的排列检验 (permutation test)计算观察到的富集得分(ES)出现的可能性。若样品量少,也可基于基因集做排列检验 (permutation test),计算
p-value。多重假设检验校正。首先对每个基因子集
s计算得到的ES根据基因集的大小进行标准化得到Normalized Enrichment Score (NES)。随后针对NES计算假阳性率。(计算NES也有另外一种方法,是计算出的ES除以排列检验得到的所有ES的平均值)Leading-edge subset,对富集得分贡献最大的基因成员。
本文通过总结多人学习使用过程中遇到的问题进一步记录软件操作过程和结果解读,力求讲清每个需要注意的细节点。
从前文中我们了解到GSEA分析的目的是要判断S集基因(基于先验知识的基因注释信息,某个关注的基因集合)中的基因是随机分布还是聚集在排序好的L基因集的顶部或底部(这便是富集分析)。
与GO富集分析的差异在于GSEA分析不需要指定阈值(p值或FDR)来筛选差异基因,我们可以在没有经验存在的情况下分析我们感兴趣的基因集,而这个基因集不一定是显著差异表达的基因。GSEA分析可以将那些GO/KEGG富集分信息中容易遗漏掉的差异表达不显著却有着重要生物学意义的基因包含在内。
下面来看看软件具体操作和结果解读。
一、软件安装
软件下载地址:http://software.broadinstitute.org/gsea/downloads.jsp
使用官方推荐的第一个软件javaGSEA Desktop Application,根据分析数据的大小和电脑内存多少可以选择下载不同内存版本的软件。该软件是基于java环境运行的,而且需要联网。若会出现打不开的现象(小编就是就碰到了),要么是没有安装java,要么是java版本太低了,安装或更新下java就能打开。也可能是网速太慢,或Java安全性问题,这时选择官网提供的第二个软件javaGSEA Java Jar file,同样依赖java运行,但不需联网,启动快。

软件启动界面如下:

二、数据准备
所有矩阵的列以tab键分割,不同类型的数据格式和后缀要求见下表。

| Data File | Content | Format | Source |
|---|---|---|---|
| Expression dataset | Contains features (genes or probes), samples, and an expression value for each feature in each sample. Expression data can come from any source (Affymetrix, Stanford cDNA, and so on). | res, gct, pcl, or txt | You create the file. 一般的基因表达矩阵整理下格式就可以。如果是其它类型数据或自己计算rank也可以,后面有更多示例。(如果后缀为txt格式,传统的基因表达矩阵就可以,第一列为基因名字,名字与待分析的功能注释数据集一致,同为GeneSymbol或EntrezID或其它自定义名字,第一行为标题行,含样品信息。gct文件需要符合下面的格式要求。) |
| Phenotype labels | Contains phenotype labels and associates each sample with a phenotype. | cls | You create the file or have GSEA create it for you. 一般是样品分组信息或样品属性度量值或时间序列信息。 |
| Gene sets | Contains one or more gene sets. For each gene set, gives the gene set name and list of features (genes or probes) in that gene set. | gmx or gmt | You use the files on the Broad ftp site, export gene sets from the Molecular Signature Database (MSigDb) or create your own gene sets file. 欲检测是否富集的基因集列表。注意基因ID与表达矩阵基因ID一致。自己准备的基因集注意格式与官网提供的gmt格式一致。 |
| Chip annotations | Lists each probe on a DNA chip and its matching HUGO gene symbol. Optional for the gene set enrichment analysis. | Chip | You use the files on the Broad ftp site, download the files from the GSEA web site, or create your own chip file. 主要是为芯片探针设计的转换文件。如果表达矩阵的基因名与注释集基因名一致,不需要这个文件。 |
1. 表达数据集文件
GESA提供有Example Datasets,下载地址:http://software.broadinstitute.org/gsea/datasets.jsp。
在这里可以下载表达矩阵Expression dataset(gct文件,常见txt格式也可以)和样品分组信息Phenotype labels(cls文件)
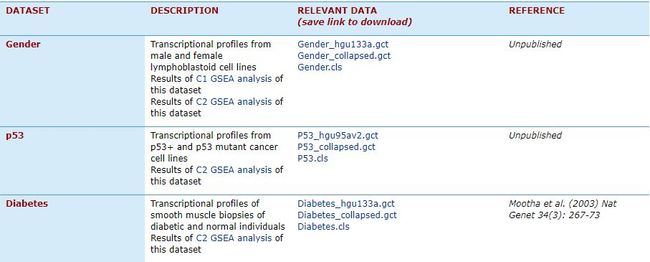
数据示例中两个gct文件都是表达矩阵,其中*hgu133a.gct文件第一列是探针名字,*collapsed.gct文件的第一列是gene symbol。
第一行:
#1.2,表示版本号,自己准备文件时照抄就行;第二行:两个数分别表示gene NAME的数量和样本数量(矩阵列数
-2);矩阵:第一列是
NAME;第二列Description,没有的话可以全用na或任意字符串填充;后面的就是基因在不同样本中标准化后的表达数据了 (部分统计量metrics for ranking genes计算需要log转换后的数据,后面会有提及。其它情况是否为log转换的数据都可用,GSEA关注的是差异,只要可比即可)。

2. 样品分组信息
第一行:三个数分别表示:34个样品,2个分组,最后一个数字1是固定的;
第二行:以
#开始,tab键分割,分组信息(有几个分组便写几个,多个分组在比较分析时,后面需要选择待比较的任意2组);(样品分组中
NGT表示正常耐糖者,DMT表示糖尿病患者,自己使用时替换为自己的分组名字)第三行:样本对应的组名。样本分组信息的第三行,同一组内的不同重复一定要命名为相同的名字,可以是分组的名字。例如相同处理的不同重复在自己试验记录里一般是Treat6h_1、Treat6h_2、Treat6h_3,但是在这里一定都要写成一样的值
Treat6h。与表达矩阵的样品列按位置一一对应,名字相同的代表样品属于同一组。如果是样本分组信息,上图中的0和1也可以对应的写成NGT和DMT,更直观。但是,如果想把分组信息作为连续表型值对待,这里就只能提供数字。

3. 功能基因集文件(gene sets)
GSEA官网提供了8种基因分类数据库,都是关于人类的数据,包括Marker基因,位置临近基因,矫正过的基因集,调控motif基因集,GO注释,癌基因,免疫基因,最新一次更新是在2018年7月,下载地址:http://software.broadinstitute.org/gsea/downloads.jsp#msigdb。

官网提供的gmt文件有两种类型,*.symbols.gmt中基因以symbols号命名,*.entrez.gmt中基因以entrez id命名。注意根据表达矩阵的基因名字命名方式选择合适的基因集。表达数据和通路数据能关联在一起依赖的是基因名字相同,所以一定保证基因命名方式的统一。

gmt格式是多列注释文件,第一列是基因所属基因集的名字,可以是通路名字,也可以是自己定义的任何名字。第二列,官方提供的格式是URL,可以是任意字符串。后面是基因集内基因的名字,有几个写几列。列与列之间都是TAB分割。
Pathway_description Anystring Gene1 Gene2 Gene3
Pathway_description2 Anystring Gene4 Gene2 Gene3 Gene5GSEA官网只提供了人类的数据,但是掌握了官网中基因表达矩阵和注释文件的数据格式,就可以根据自己研究的物种,在公共数据库下载对应物种的注释数据,自己制作格式一致的功能基因集文件,这样便就可以做各种物种的GSEA富集分析了。
4. 芯片注释文件
如果分析的表达数据是芯片探针数据就需要用到芯片注释文件(chip),用来做ID转换,把探针名字转换为基因名字。如果我们的表达数据文件中已经是基因名了就不再需要这个文件了。
三、分析参数设置和软件运行
演示使用的数据来自GSEA官网:
表达矩阵:Diabetes_collapsed_symbols.gct
样品分组信息:Diabetes.cls
基因功能分类数据选择GO数据库:c5.all.v6.2.symbols.gmt
因为表达矩阵与注释中基因名字可以直接对应,第四个文件不需要
1. 数据导入
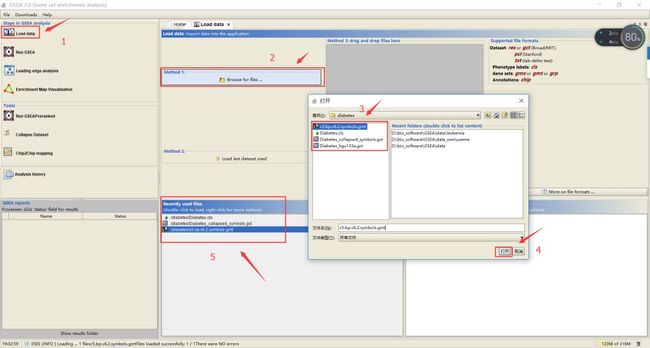
按照上图步骤依次点击Load data——Browse for file——在弹出文件框中找到待导入的文件,选中点击打开即可;
若文件格式没问题会弹出一个提示There were no error的框,证明文件上传成功,并且会显示在5所示的位置;若出错,请仔细核对文件格式。
注意:1)本地文件存放路径不要有中文、空格(用_代替空格)和其他特殊字符;2)所有用到的文件都需要通过上述方式先上传至软件;3)数据上传错误后可以通过点击工具栏file——clear recent file history进行清除。
2. 指定参数
点击软件左侧Run GSEA,将跳出参数选择栏。参数设置分为三个部分Require fields(必须设置的参数项)、Basic fields(基本参数设置栏)和Advanced fields(高级参数设置栏),后面两栏的参数一般不做修改,使用默认的就行。后面两部分参数设置,如果涉及到需要根据实验数据做调整的地方,会在后面的分析中会提到。
1)Require fields

Expression dataset: 导入表达数据集文件,点击后自动显示上一步中从本地导入软件内的文件,所以一定要确认上一步导入数据是否成功;
Gene sets database: 基因功能集数据库,可以从本地导入(上一步);
在联网的情况下软件也可以为自动下载GSEA官网中的
gene sets文件;Number of permutations: 置换检验的次数,数字越大结果越准确,但是太大会占用太多内存,软件默认检验1000次。
软件分析时会得到一个基因富集的评分(ES),但是富集评分是否具有统计学意义,软件就会采用随机模拟的方法,根据指定参数随机打乱1000次,得到1000个富集评分,然后判断得到的ES是否在这1000个随机产生的得分中有统计学意义。测试使用时建议填一个很小的数如
10,先让程序跑通。真正分析时再换为1000。Phenotype labels: 选择比较方式,如果文件只有2个组别的话就比较方便了,任意选一个就行,哪个在前在后全在自己怎么解释方便;如果数据有多组的话,GSEA会提供两两间比较的组合选项或者某一组与剩下所有组的比较。选择好后,GSEA会在分析过程中根据组别信息自动到表达数据集文件中提取对应的数据作比较。
Collapse dataset to gene symbols: 如果表达数据集文件中NAME已经与gene sets database中名字一致,选择
FALSE,反之选择TRUE。Permutation type: 选择置换类型,
phenotype或者gene sets。每组样本数目大于7个时 ,建议选择phenotype,否则选择gene sets。
Chip platform: 表达数据集为芯片数据时才需要,目的是对ID进行注释转换,如果已经转换好了就不需要了。应该也适用于其它需要转换ID的情况,不过事先转换最方便。
2)Basic fields

通常选择默认参数即可,在此简单介绍一下
Analysis name: 取名需要注意不能有空格,需要用
_代替空格。如果做的分析多,最好选择一个有意义的名字,比如shengxinbaodian(生信宝典全拼),方便查找。Enrichment statistic: 基因集富集分析(PNAS)的最后一部分给出了GSEA中所用方法的数学描述,感兴趣的可以查看一下论文。在此给出每种富集分析不同算法的参数情况:▪ classic: p=0 若基因存在,则ES值加1;若基因不存在,则ES值减1 ▪ weighted (default): p=1 若基因存在,则ES加rank值;若基因不存在,则ES减rank值 ▪ weighted_p2: p=2 基因存在,ES加rank值的平方,不存在则减rank值的平方 ▪ weighted_p1.5: p=1.5 基因存在,ES加rank值得1.5次方,不存在则减rank值得1.5次方
备注:如果想用其它加权,就自己计算rank值,使用preranked mode。
Metric for ranking genes: 基因排序的度量
下面提到的
均值也可以是中位数。如果表型是分组信息,GSEA在计算分组间的差异值时支持5种统计方式,分别是
signal2noise、t-Test、ratio_of_class、diff_of_class(log2转换后的值计算倍数)和log2_ratio_of_class。下面公式很清楚。
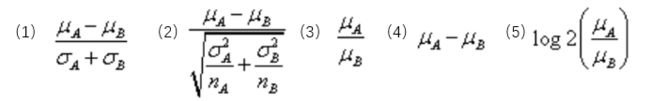
如果表型是连续数值信息(定量表型): GSEA通过表型文件(
cls)和表达数据集文件(gct),使用pearson相关性、Cosine、Manhattan或Euclidean指标之一计算两个配置文件之间的相关性。(注意:若是分组表型文件想转换为定量表型,cls文件中分类标签应该指定为数字)
Gene list sorting mode: 对表达数据集中的基因进行排序,按照排序度量的真实值(默认)或者绝对值排序;
Gene list ordering mode: 使用此参数确定表达数据集中基因是按照降序(默认)或者升序排列;
Max size & Min size: 从功能基因集中筛选出不属于表达数据集中的基因后,剩下基因总数在此范围内则保留下来做后续的分析,否则将此基因集排除;一般太多或太少都没有分析意义。
Save results in this folder: 在此可以选择分析文件在本地电脑的存储地址。
3)Advanced fields

Collapsing mode for probe sets => 1 gene:多个探针对应一个基因时的处理方式。
Normalized mode: 富集得分的标准化方式。
Randomization mode:只用于phenotype permutation。
Median for class metrics: 计算metrics ranking时用中值而不是平均值。
Number of markers:红蝶图中展示的Gene Marker数目。
Plot graphs for the top sets of each phenotype:绘制多少GSEA plot,默认top 20,其它不绘制。一般会把这个值调高。
Seed for permutation:随机数种子,如果想让每次结果一致,这里需要设置同样的一个整数。
以上参数都设置好后点击参数设置栏下方的一个绿色按钮Run,若软件左下方GSEA reports处的状态显示Running的话则表示运行成功,此过程大概需要十分钟左右,视数据大小而定。

Command:显示运行这个分析的命令行,以后就可以批量运行类似分析了。
四、结果解读
数据分析完后的结果会保存到我们设置的路径下,点开文件夹中的index.html就可以查看网页版结果,更加方便。
结果报告分为多个子项目,其中最重要的是前面两部分,基因富集结果就在这里。从第三部分开始其实是软件在分析数据的过程产生的中间文件, 也很重要,读懂后可以加深对GSEA分析的认识,理解我们是如何从最初的基因表达矩阵得到最终的结果(即报告的前两个项目)。建议先从Dataset details看起,然后再返回看第一部分的结果报告。
1. Enrichment in phenotype
以正常人组NGT的17个样本数据为例解析最终结果。

报告首页文字总结信息表示:
经过条件筛选后还剩下3953个GO条目,其中1697个GO条目在
NGT组中富集;有36个GO基因条目在FDR<25%的条件下显著富集,这部分基因最有可能用于推进后续实验;
在统计检验
p<0.01,p<0.05的条件下分别有19和114个GO条目显著富集;结果有多种显示方式:图片快照(snapshot)、网页(html)和表格(Excel)形式;
点击
Guide to可以查看官方帮助解读结果的文档。
1) 点击enrichment results in html,在网页查看富集结果,如下:
GS:基因集的名字,GO条目的名字
SIZE:GO条目中包含表达数据集文中的基因数目(经过条件筛选后的值);
ES:富集评分;
NES:校正后的归一化的ES值。
由于不同用户输入的基因数据库文件中的基因集数目可能不同,富集评分的标准化考虑了基因集个数和大小。
其绝对值大于
1为一条富集标准。计算公式如下:

NOM p-val:即
p-value,是对富集得分ES的统计学分析,用来表征富集结果的可信度;FDR q-val:即
q-value,是多重假设检验校正之后的p-value,即对NES可能存在的假阳性结果的概率估计,因此FDR越小说明富集越显著;RANK AT MAX:当ES值最大时,对应基因所在排序好的基因列表中所处的位置;(注:GSEA采用p-value<5%,q-value<25%进行数据过滤)
LEADING EDGE:该处有3个统计值,
tags=59%表示核心基因占该基因集中基因总数的百分比;list=21%表示核心基因占所有基因的百分比;signal=74%,将前两项统计数据结合在一起计算出的富集信号强度,计算公式如下: 其中n是列表中的基因数目,nh是基因集中的基因数目
其中n是列表中的基因数目,nh是基因集中的基因数目
点击Details跳转至对应的详情结果。只有前20个GO富集详情可以查看,想要生成的结果报告可以查看更多的富集信息,可以通过在Advanced fields处设置参数Plot graphs for the top sets of each phenotype。
2)Details for gene set 首先是一个选定GOset下的汇总信息表,每一部分意思在上面已做解释,其中
首先是一个选定GOset下的汇总信息表,每一部分意思在上面已做解释,其中Upregulated in class表示该基因集在哪个组别中高表达,这个主要看富集分析后的leading edge分布位置。
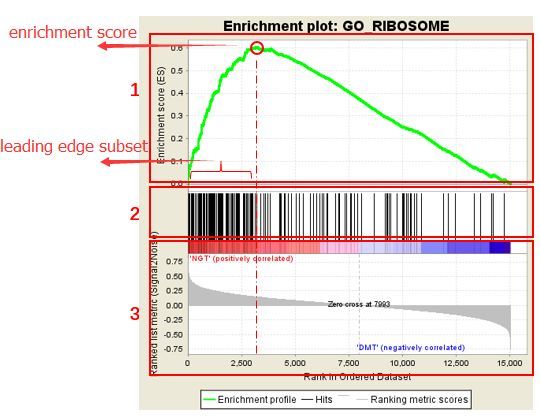 接下来是富集分析的图示,该图示分为三部分,在图中已做标记:
接下来是富集分析的图示,该图示分为三部分,在图中已做标记:
第一部分是
Enrichment score折线图:显示了当分析沿着排名列表按排序计算时,ES值在计算到每个位置时的展示。最高峰处的得分 (垂直距离0.0最远)便是基因集的ES值。第二部分,用线条标记了基因集合中成员出现在基因排序列表中的位置,黑线代表排序基因表中的基因存在于当前分析的功能注释基因集。leading edge subset 就是(0,0)到绿色曲线峰值ES出现对应的这部分基因。
第三部分是排序后所有基因rank值得分布,热图红色部分对应的基因在
NGT中高表达,蓝色部分对应的基因在DMT中高表达,每个基因对应的信噪比(Signal2noise,前面选择的排序值计算方式)以灰色面积图显展示。
在上图中,我们一般关注ES值,峰出现在排序基因集的前端还是后端(ES值大于0在前端,小于0在后端)以及Leading edge subset(即对富集贡献最大的部分,领头亚集);在ES图中出现领头亚集的形状,表明这个功能基因集在某处理条件下具有更显著的生物学意义;对于分析结果中,我们一般认为|NES|>1,NOM p-val<0.05,FDR q-val<0.25的通路是显著富集的。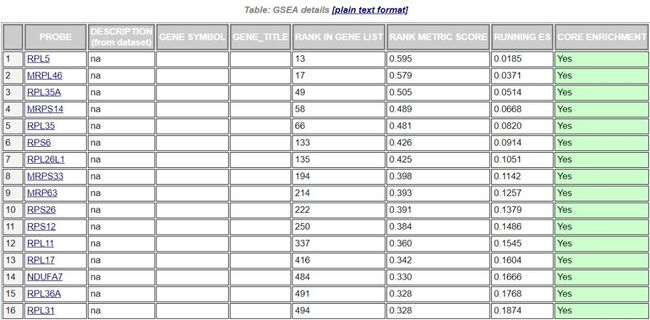 最后还有一个该GO基因集下每个基因的详细统计信息表,
最后还有一个该GO基因集下每个基因的详细统计信息表,RANK IN GENE LIST表示在排序好的基因集中所处的位置;RANK METRIC SCORE是基因排序评分,我们这里是Signal2noise;RUNNING ES是分析过程中动态的ES值;CORE ENRICHMENT是对ES值有主要贡献的基因,即Leading edge subset,在表中以绿色标记。
2. Dataset details
芯片原始数据和去重后的数据;如果分析的时候没有用到芯片数据或没涉及到名字转换则前后基因数目一样。
3. Gene set details
我们分析提供的gmt文件中有多个GO条目,每个GO条目里又有多个基因;GSEA分析软件会在每个GO条目中搜索表达数据集gct文件中的基因,并判断有多少个在GO条目中;若经过筛选后保留在GO条目中的基因在15-500(闭区间)时该GO条目才被保留下来进行后续的分析。

此结果显示我们从5917个GO条目中淘汰了了1964个GO,剩下3953个GO条目用作后续分析。
点击gene sets used and their sizes可以下载详细Excel表。
Excel第一列是GO名称,第二列是GO条目中包含的基因数目,第三列是筛选后每个GO中还有多少基因属于表达数据集文件中的基因,不满足参数(15-500)的条目被抛弃,显示为Rejected不纳入后续分析。
备注: 此处的筛选范围15-500是可调参数,在软件的参数basic fields处的Max size和Min size处更改。
4. Gene markers for the NGT versus DMT comparison
这部分展示的是我们提供的表达数据集文件中的基因在两个组别中的表达情况。
输入的文件中总共有15056个基因,其中有7993个基因在正常人(NGT)中表达更高,占总基因数的53.1%;有7063个基因在糖尿病患者(DMT)中表达更高,占总基因数的46.9%。后面一个面积百分比,稍后看图的时候再做解释。

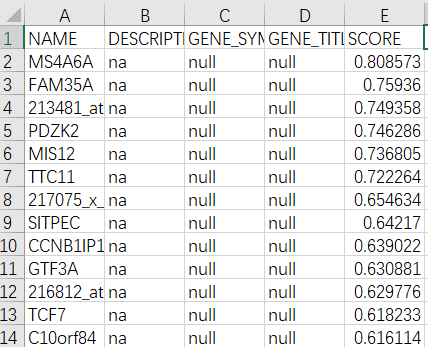
点击rank ordered gene list可以下载一个排序好的基因集Excel表,排序原则是根据Basic fields参数设置处的Metric for ranking genes决定的。我们选的是信噪比(signal2noise),显示在表格中的最后一列。根据NGT_vs_DMT评分得到一个降序排列的基因集,之后便可以做基因的富集分析了。
GSEA基因富集分析的原理就是基于该排列好的基因集,从第一个基因开始判断该基因是否存在于经过筛选的GO功能基因集中,如果存在则加分,反之减分。所以评分过程是一个动态的过程,最终我们会得到一个评分峰值,那就是GO功能富集的评分。加分规则通过Basic fields参数设置处的Enrichment statistic决定的。
接着有一个分析的结果的热图和gene list相关性的图。
热图中展示了分别在两组处理中高表达的前50个基因,总共100个基因的表达情况。
gene list相关性图如下。横坐标是已经排序好的基因,纵坐标是signal2noise的值。虚线左侧的基因是在NGT中高表达,右侧的基因在DMT中高表达。这部分结果报告中的面积比就是基于该图计算的,可以看出面积百分比和基因数目百分比有一定的差异,面积百分比可以从整体上反映组间信噪比的大小。
Butterfly plot显示了基因等级与排名指标评分之间的正相关(左侧)和负相关性(右侧)。左侧蓝色虚线和右侧红色虚线是真实的信噪比结果,其他颜色的线是软件对数据做了随机重排后的结果。默认情况下,图形只显示前100个基因,也就是排名第一和最后100个基因。可以使用运行GSEA页面上Advanced fields处的Number of markers来更改显示的基因数量。

5. Global statistics and plots
这部分包含两个图:1) p值与归一化富集分数(NES)的对比图,这提供了一种快速、直观的方法来掌握有意义的富集基因集的数量。2) 通过基因集的富集分数统计图,提供了一种快速、直观的方法来掌握富集的基因集的数量。


理解了上面各个部分的结果后,再回过头看这张GSEA分析原理图就简单了。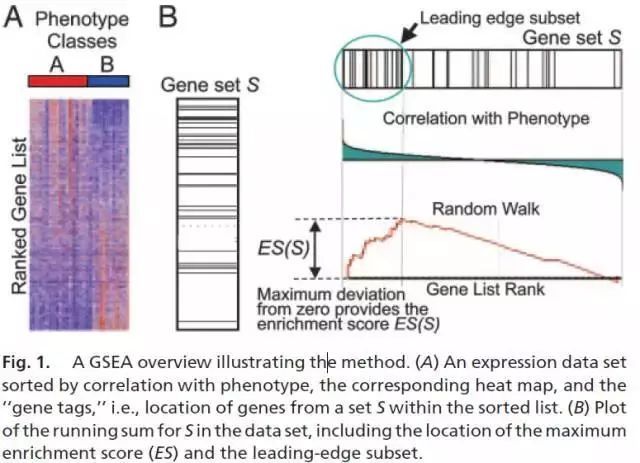
Cytoscape富集网路可视化
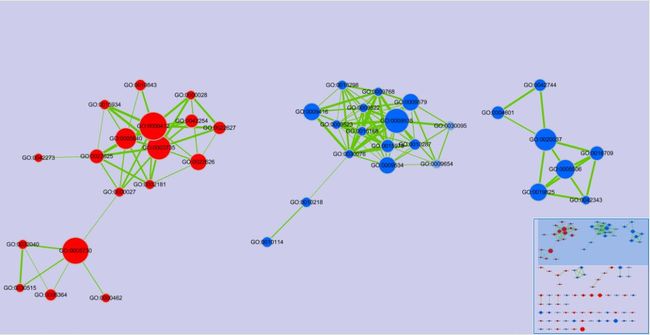
在GSEA软件的左侧提供了Enrichment Map Visualization的功能,点击后GSEA软件会自动调用Cytoscape,建议等待Cytoscape启动后再进行接下来的操作,且保证在分析过程中Cytoscape是处于开启状态。
选择一个GSEA分析结果,点击Load GSEA Results,其他项为默认值就行,点击Build Enrichment Map以展示基因富集结果的网络图。(备注:GSEA分析结果用的是和上面演示数据不同的文件,可自行更改)

运行成功之后会弹出下面的提示框,结果直接展现在了Cytoscape中,如下图所示:

Graphpad作图比较多个ES
GSEA富集分析可视化结果是给每个功能基因集富集情况单独出一张图,有的时候我们想要比较基因集在两个不同的GO中的富集情况,利用GSEA软件分析得到的Excel结果表,提取有用的数据结果,在graphpad里进行加工再出图,可以达到我们想要的结果!
效果图如下: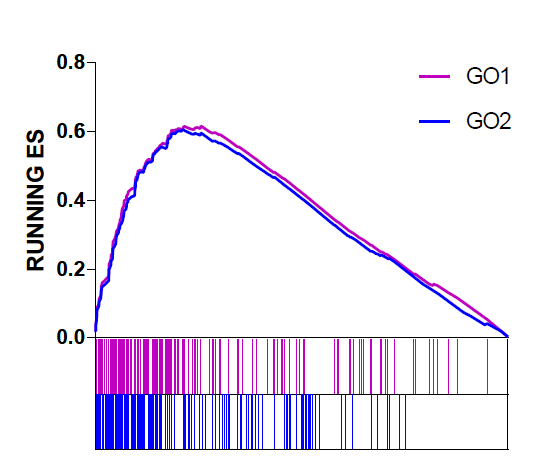
《Graphpad,经典绘图工具初学初探》一文中介绍了graphpad入门的基础知识,基本操作可以单击回看。最近使用graphpad发现其多图排版功能十分强大,不仅可以实现多个图形排版还能实现图层叠加。上面这个图的作图思路也就是把该图拆分为两部分,Enrichment score和基因位置分布条带图。
在GSEA分析结果文件夹里随便找一个感兴趣的GO条目分析结果Excel表,作图需要提取的信息即图中标黄的部分,RANK IN GENE LIST和RUNNING ES。

加工一下已有数据,添加一列high取值都为0.1,设置高度,黄色部分的数据就是用来绘制基因位置分布条带图的;绿色部分用来绘制动态的ES评分曲线。
打开graphpad之后,我们在XY类图下选择Enter and plot a single Y value for each point,将两部分数据分开粘贴到软件不同数据表格中(如下图左侧所示),下图中间展示两个图选择的不同绘图方式,调整参数后最终得到右侧的结果。

在左侧目录树处点击layout创建一个图形排版界面,将Graphs下的图形复制粘贴到layout1下,拖拉移动位置很快就能将两部分图对齐。
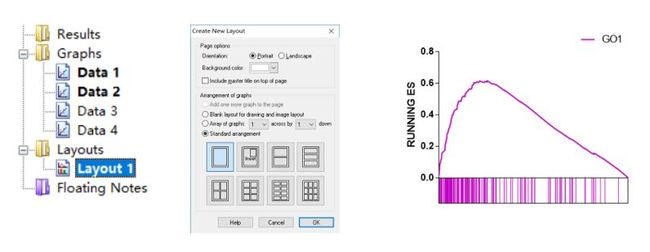 之后用同样地方式画另外一个富集结果,粘贴到layout1中便得到最开始展示的图。
之后用同样地方式画另外一个富集结果,粘贴到layout1中便得到最开始展示的图。
注意:设置X轴的范围是1到总排序基因数,Y轴是0到多个富集分析得分的最大值。
GO、GSEA富集分析一网打进
GSEA富集分析 - 界面操作
无需写代码的高颜值富集分析神器
去东方,最好用的在线GO富集分析工具
没钱买KEGG怎么办?REACTOME开源通路更强大
超简便的国产lncRNA预测工具LGC
我想做信号通路分析,但我就是不想学编程
这个只需一步就可做富集分析的网站还未发表就被CNS等引用超过350次
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|




























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集


