- 2025.03.22【读书笔记】| fastq-multx:高效barcode拆分数据解决工具
穆易青
读书笔记数据处理读书笔记linux运维服务器
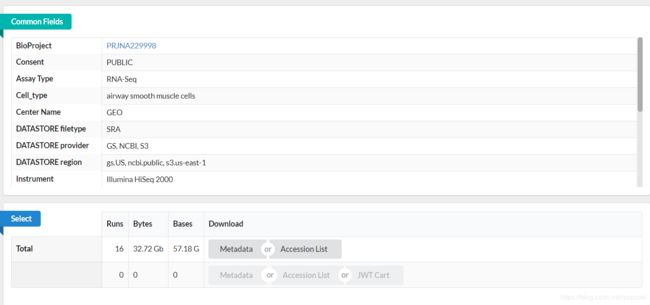
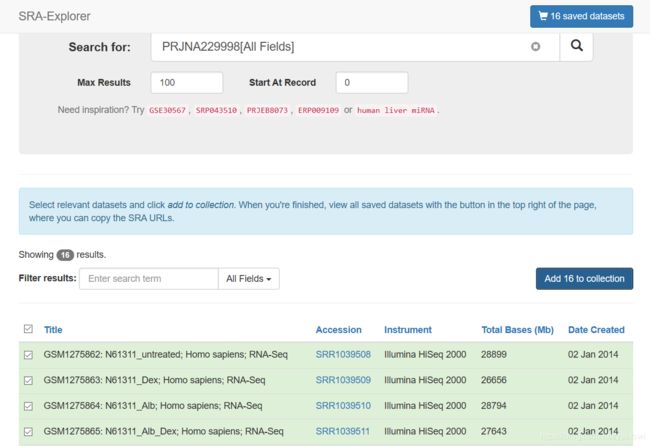
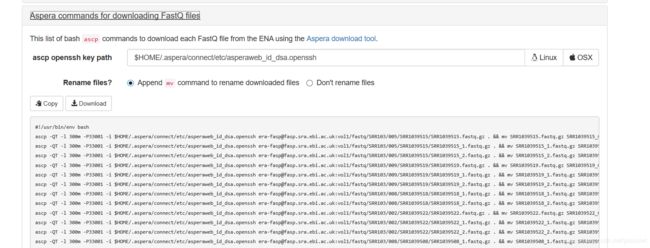
文章目录1.工具介绍为什么需要`fastq-multx`?`fastq-multx`的特点2.安装方式通过源代码编译安装使用包管理器安装3.使用命令基本命令高级参数设置结语1.工具介绍在生物信息学的世界里,工具的选择至关重要。今天,我们要介绍的这个工具,就是fastq-multx,一个用于高效barcode去复用和demultiplex的解决方案。fastq-multx是一个专门设计用于处理高通量
- 数学领域的跨时代进化与升级:从公理化到智能化的破茧之路
夏末之花
算法
作者:夏末之花|发布时间:2025-03-16|阅读量:10万+|点赞数:5.6万引言:数学的“破茧时刻”与文明跃迁人类历史上,数学的每一次重大突破都像一次“破茧时刻”,推动文明跨越式发展。从古希腊的几何公理化到牛顿的微积分,再到20世纪的计算机理论,数学始终是科学革命的基石。而在21世纪的今天,随着量子计算、人工智能、生物信息等技术的爆发,数学正迎来新一轮的进化与升级——从纯粹的逻辑工具,演变为
- R语言绘图 | 环状柱状图+散点柱状组合图绘制
小杜的生信筆記
R语言精美图形绘制教程r语言开发语言科研绘图生物信息学
原文:R语言绘图|环状柱状图+散点柱状组合图绘制(点击访问)小杜的生信筆記,主要发表或收录生物信息学教程,以及基于R分析和可视化(包括数据分析,图形绘制等);分享感兴趣的文献和学习资料!!
- 计算基因组学需要计算机知识吗,生物信息学——计算基因组学的一些参考书
weixin_39610422
计算基因组学需要计算机知识吗
有两个都可以在新浪爱问资料Bioinformatics.For.Dummies.2nd.Ed.2007.pdfAnIntroductiontoBioinformaticsAlgorithms.pdf另外看到Virginia大学的一些课程The2012ComputationalGenomicsCoursehasbeenrescheduledtoNovember28-December4,2012用mo
- Python在生物信息学中的应用:基因组学与蛋白质组学
PyTechShare
Python教程-基础python
摘要:本文主要介绍了Python在生物信息学中的应用,特别是在基因组学和蛋白质组学领域。文章详细讲述了各个原理,并以代码实例展示了实际应用。我们将探讨如何利用Python分析基因组数据,解析蛋白质序列,以及进行比对分析等。文章目录1.引言2.分析基因组数据2.1读取和解析FASTA文件2.2基因频率分析2.3代码实例3.蛋白质组学3.1解析蛋白质序列3.2蛋白质序列比对3.3代码实例4.总结1.引
- 数组中最长递增子序列问题的深入研究
cloudman08
算法
目录摘要一、引言二、问题定义三、问题分析3.1暴力枚举法的困境3.2动态规划的应用3.3二分查找优化四、算法设计4.1动态规划算法4.2二分查找优化算法4.3代码实现(Python)4.4代码解释五、复杂度分析5.1动态规划算法复杂度5.2二分查找优化算法复杂度六、实际应用6.1数据分析6.2生物信息学6.3信号处理七、结论摘要在数组处理的算法领域,寻找最长递增子序列是一个经典且具有广泛应用的问题
- 基于多模态大模型的不完整多组学数据特征选择策略
m0_65156252
人工智能
基于多模态大模型的不完整多组学数据特征选择策略是当前生物信息学和精准医学领域的一个前沿问题。在多组学数据中,通常包括不同层次的生物信息(如基因组、转录组、蛋白质组、代谢组等),这些数据通常存在缺失、噪声或不一致的情况。因此,如何有效地在这些不完整的数据中进行特征选择,是实现精确疾病预测和个性化治疗的关键。结合多模态大模型(如自监督学习、图神经网络、Transformer等)可以有效解决这一问题。以
- 生物信息学工作流(Bioinformatics Workflow):概念、历史、现状与展望?
lisw05
生物信息学生物信息学工作流
李升伟整理1.引言生物信息学工作流是指通过一系列计算步骤和工具,对生物学数据进行处理、分析和解释的系统化流程。随着高通量测序技术的普及和生物数据的爆炸式增长,生物信息学工作流在基因组学、转录组学、蛋白质组学等领域中扮演着至关重要的角色。它不仅提高了数据分析的效率,还为生命科学研究提供了新的视角和方法。2.生物信息学工作流的概念生物信息学工作流的核心是将复杂的生物学数据分析任务分解为多个可管理的步骤
- LM_Funny-2-01 递推算法:从数学基础到跨学科应用
王旭·wangxu_a
算法
目录第一章递推算法的数学本质1.1形式化定义与公理化体系定理1.1(完备性条件)1.2高阶递推的特征分析案例:Gauss同余递推4第二章工程实现优化技术2.1内存压缩的革新方法滚动窗口策略分块存储技术2.2异构计算加速方案GPU并行递推量子计算原型第三章跨学科应用案例3.1密码学中的递推构造混沌流密码系统3.2生物信息学的序列分析DNA甲基化预测第一章递推算法的数学本质1.1形式化定义与公理化体系
- R语言安装生物信息数据库包
Bio Coder
R语言r语言数据库
R语言安装生物信息数据库包在生物信息学领域,R语言是重要的数据分析工具。今天,我们就来聊聊在R语言环境下,安装生物信息数据库包(org.*.*.db)的步骤。为什么要安装org.*.*.db系列包生物信息学分析中,我们常处理基因相关数据,比如基因功能注释、位置、参与的生物学通路等。org.*.*.db系列包就像基因百科全书,提供不同物种的基因注释信息。比如研究人类基因时,能帮我们快速获取基因别名、
- 生物信息数据库开发之单细胞数据库scrna db(一)
北京生信课堂
数据库生物信息学r语言python
单细胞数据库构建优质已整合的单细胞数据库背景知识需求分析数据库类型数据库构建过程优质已整合的单细胞数据库如果读者只想获得一个现成的内容丰富的单细胞数据库加入至自己的PC或linux服务器,可以跳过下面的详细理论教程数据库下载链接:点击下载单细胞数据库。包含约800个细胞数据的中小型数据库,维度约为20000x800,部分为作者公司数据,部分为国际数据库数据,包含T-cell,B-cell,NK-c
- 基于python使用scanpy分析单细胞转录组数据
探序基因
单细胞分析python开发语言
探序基因肿瘤研究院整理相关后缀的格式介绍:.h5ad:是一种用于存储单细胞数据的文件格式,可以通过anndata库在Python中处理.loom:高效的数据存储格式(.loom文件),使得用户可以轻松地存储、查询和分析大规模的单细胞数据集。Loompy的设计目标是提供一个快速、灵活且易于使用的工具,以支持生物信息学家和研究人员在单细胞水平上进行数据分析。python的单细胞转录组数据结构说明:da
- Ensembl ID转Gene Symbol
Red Red
生信小技巧r语言数据库
生物信息中EnsemblID转换为GeneSymbolTCGA数据库该方法比较简单,无需安装过多的包,因为安装了几个包显示和R版本不兼容,浪费很多时间,最后用这个包成功将Ensemble转换成IDR:#加载org.HS.eg.dblibrary(org.Hs.eg.db)#获取所有的ENSEMBLID到k中k=keys(org.Hs.eg.db,keytype="ENSEMBL")k#找到ENSE
- Perl 语言入门学习指南:探索高效脚本编程的奥秘
我的运维人生
简约运维perlPerl编程脚本语言文本处理Perl基础语法
引言Perl,全称PracticalExtractionandReportLanguage,是一种功能强大的编程语言,特别擅长于文本处理、报告生成以及系统自动化管理任务。自1987年诞生以来,Perl凭借其灵活性、强大的内置功能库和广泛的社区支持,在Web开发、生物信息学、网络管理等多个领域发挥着重要作用。本文旨在为初学者提供一份Perl语言入门学习指南,帮助大家快速掌握这门强大的脚本语言。一、P
- gseapy python包GO、KEGG富集(注释)分析
loong_XL
生信pythongolang开发语言
文档案例参考:https://gseapy.readthedocs.io/en/latest/gseapy_example.html#Over-representation-analysis-by-Enrichr-web-services简介:富集分析是一种常见的生物信息学分析方法,通过比较一个给定的基因集(如一组显著差异表达基因)与已知的生物过程、通路或功能的数据库,来发现哪些过程、通路或功能与
- Java 大视界 -- Java 大数据在生物信息学中的应用与挑战(67)
青云交
大数据新视界Java大视界大数据生物信息学基因序列分析蛋白质结构预测数据质量计算资源机器学习
亲爱的朋友们,热烈欢迎来到青云交的博客!能与诸位在此相逢,我倍感荣幸。在这飞速更迭的时代,我们都渴望一方心灵净土,而我的博客正是这样温暖的所在。这里为你呈上趣味与实用兼具的知识,也期待你毫无保留地分享独特见解,愿我们于此携手成长,共赴新程!一、欢迎加入【福利社群】点击快速加入:青云交灵犀技韵交响盛汇福利社群点击快速加入2:2024CSDN博客之星创作交流营(NEW)二、本博客的精华专栏:大数据新视
- 因果推断与机器学习—因果表征学习与泛化能力
樱花的浪漫
因果推断机器学习学习人工智能深度学习自然语言处理计算机视觉
近十年来,深度学习在多个领域取得了巨大成功,包括机器视觉、自然语言处理、语音识别和生物信息等。这些成功为机器学习技术的进一步发展和应用奠定了基础。表征学习是深度学习的核心技术之一。在机器学习问题中,其主要目的是从观测到的低级变量中提取信息,进而学习到能够准确预测目标变量的高级变量。这种从低层次到高层次变量的学习过程,有助于模型更好地理解数据和进行预测。以德国马克斯-普朗克研究所的BernhardS
- 《AI赋能行业实战:揭秘企业数字化转型最佳实践,落地案例深度解析!》 ---- 总目录
shiter
人工智能系统解决方案与技术架构人工智能大数据AI
文章大纲金融行业落地实践浅析基于PySpark进行信用卡评分--实战案例迁移学习小样本金融风控生物信息识别大健康行业落地实践浅析传统行业深度融合升级如何深度参与创业?物联网行业案例浅析智慧园区案例浅析计算机视觉应用案例计算机视觉入门学习国外的资源国内的资源YOLO学习modelzoo计算机视觉基础目标检测YOLOv5YOLOv8自动缺陷检测(AutoDefectClassification)、零件
- 三甲医院大型生信服务器多配置方案剖析与应用(2024版)
Allen_LVyingbo
数智化医院2024服务器数据库运维
一、引言1.1研究背景与意义在当今数智化时代,生物信息学作为一门融合生物学、计算机科学和信息技术的交叉学科,在三甲医院的科研和临床应用中占据着举足轻重的地位。随着高通量测序技术、医学影像技术等的飞速发展,生物医学数据呈爆发式增长,这些数据涵盖了基因组、蛋白质组、代谢组等多个层面的信息,为医学研究和临床诊断提供了前所未有的机遇与挑战。从科研角度来看,生物信息学助力三甲医院开展前沿性的医学研究。通过对
- Python3 【集合】项目实战:3 个新颖的学习案例
李智 - 重庆
Python精讲精练-从入门到实战python经验分享案例学习编程技巧
Python3【集合】项目实战:3个新颖的学习案例以下是3个应用“Python集合”知识的综合应用项目,这些项目具有新颖性、前瞻性和实用性,每个项目都包含完整的代码、解释说明、测试案例和执行结果。基因序列比对文章推荐系统运行日志分析项目1:基因序列比对(集合运算与去重)项目描述在生物信息学中,比对两个基因序列的相似性。使用集合的交集和并集计算相似度。代码实现#基因序列(简化为字符串集合)seque
- AI人工智能深度学习算法:在生物信息学中的应用
AI大模型应用之禅
AI大模型与大数据计算科学神经计算深度学习神经网络大数据人工智能大型语言模型AIAGILLMJavaPython架构设计AgentRPA
AI人工智能深度学习算法:在生物信息学中的应用关键词:人工智能、深度学习、生物信息学、基因组学、蛋白质结构预测、药物发现、个性化医疗文章目录AI人工智能深度学习算法:在生物信息学中的应用1.背景介绍2.核心概念与联系2.1人工智能(AI)2.2机器学习(ML)2.3深度学习(DL)2.4生物信息学2.5应用领域3.核心算法原理&具体操作步骤3.1算法原理概述3.1.1卷积神经网络(CNN)3.1.
- AlphaFold2的思路总结(十五)
xiaofengzihhh
蛋白质结构预测深度学习人工智能神经网络
2021SC@SDUSC这学期的代码分析工作接近尾声了,我想简单总结一下AlphaFold2的总体思路 具体来看,AlphaFold2主要利用多序列比对(MSA),把蛋白质的结构和生物信息整合到了深度学习算法中。它主要包括两个部分:神经网络EvoFormer和结构模块(Structuremodule)。一、EvoFormer 在EvoFormer中,主要是将图网络(Graphnetworks)
- Spark GraphX原理与代码实例讲解
AI大模型应用之禅
AI大模型与大数据计算科学神经计算深度学习神经网络大数据人工智能大型语言模型AIAGILLMJavaPython架构设计AgentRPA
SparkGraphX原理与代码实例讲解作者:禅与计算机程序设计艺术/ZenandtheArtofComputerProgramming1.背景介绍1.1问题的由来随着互联网和大数据技术的迅猛发展,社交网络、推荐系统、生物信息学、图分析等领域对图计算的需求日益增长。传统的图处理技术如GraphLab、Neo4j等,虽然功能强大,但往往存在扩展性差、易用性低、计算效率不足等问题。为了解决这些问题,A
- R语言的计算机基础
java熊猫
包罗万象golang开发语言后端
R语言计算机基础引言R语言是一种用于数据分析、统计计算和图形显示的编程语言。它被广泛应用于统计学、数据科学、生态学、生物信息学等多个领域。由于其强大的功能和灵活性,R语言在学术界和工业界都得到了广泛的认可和应用。本文将从R语言的基本概念、数据类型、数据结构、函数、控制结构、图形绘制等方面进行介绍,帮助读者掌握R语言的基础知识。一、R语言的基本概念R语言源于新西兰的维特利大学,最初由RobertGe
- Web APP 阶段性综述
预测模型的开发与应用研究
APPconstructionwebapp
WebAPP阶段性综述当前,WebAPP主要应用于电脑端,常被用于部署数据分析、机器学习及深度学习等高算力需求的任务。在医学与生物信息学领域,WebAPP扮演着重要角色。在生物信息学领域,诸多工具以WebAPP的形式呈现,相较之下,医学领域的此类应用数量相对较少。在医学和生物信息学的学术论文中,WebAPP是展示研究成果的有效工具,并且还能部署到网络上,服务于实际应用场景。ShinyAPP平台特性
- GEO数据的下载和处理|GEO数据转换为Gene symbol|GEO注释文件提取symbol|查看样本标签|查看GEO数据疾病或正常|生物信息基础
Red Red
生信小技巧学习笔记生物信息r语言GEO数据库数据库
GEO数据的下载和处理|GEO数据转换为Genesymbol|GEO注释文件提取symbol|查看样本标签|查看GEO数据疾病或正常|生物信息基础数据的下载和处理首先在GEO数据库中通过GSEID找到相关数据,然后下载txt文件。数据读取与处理。#设置工作路径,也就是你的分析数据存放以及要保存到地方setwd(dir="C:\\Users\\LiaoMinzhen\\PycharmProjects
- 生物信息名词汇总|生物信息基础知识
Red Red
生信小技巧学习笔记
生物信息名词汇总|生物信息基础知识GWAS-Genome-wideassociationstudies,全基因组关联研究:用于识别遗传区域(基因组)和性状/疾病之间关联的方法。Predixcan:GWAS找到大量的SNP,可是可以解释生物学功能的SNP位点却是很有限的。gene-based关联分析软件——PredicXcan。PrediXcan包括两个步骤:-在具有可用基因型的队列中预测基因表达(
- 推荐一份生物信息学入门很好的参考材料
小明的数据分析笔记本
链接是https://bioinformatics.uconn.edu/resources-and-events/tutorials-2/这个是康涅狄格大学(UniversityofConnecticut)提供的一份教程,主要的内容包括1、生物信息学中经常用到的文件格式image.png2、linux操作系统和R语言的基础知识image.png3、转录组数据的处理流程image.png这里包括有参
- 【机器学习】朴素贝叶斯方法的概率图表示以及贝叶斯统计中的共轭先验方法
Lossya
机器学习概率论人工智能朴素贝叶斯共轭先验
引言朴素贝叶斯方法是一种基于贝叶斯定理的简单概率模型,它假设特征之间相互独立。文章目录引言一、朴素贝叶斯方法的概率图表示1.1节点表示1.2边表示1.3无其他连接1.4总结二、朴素贝叶斯的应用场景2.1文本分类2.2推荐系统2.3医疗诊断2.4欺诈检测2.5情感分析2.6邮件过滤2.7信息检索2.8生物信息学三、朴素贝叶斯的优点四、朴素贝叶斯的局限性4.1特征独立性假设4.2敏感于输入数据的表示4
- 零基础入门生信数据分析——导读
呆猪儿
生信之转录组——上游分析生信之转录组——下游分析学习方法r语言数据分析数据库数据挖掘需求分析大数据
零基础入门生信数据分析——导读生信数据分析,即生物信息学数据分析,是一个涵盖了生物学、计算机科学、数学和统计学等多个领域的交叉学科。它主要利用计算机算法和统计方法对生物学数据进行处理、分析和解释,以揭示生物分子、细胞、组织和生物体等各个层次的生物学规律和机制。本帖主要是为生信数据分析的各个分析点提供跳转链接(简单说就是提供了一个目录供大家选择自己想要的知识点可以直接跳转)关联的生信数据分析的分析点
- java数字签名三种方式
知了ing
javajdk
以下3钟数字签名都是基于jdk7的
1,RSA
String password="test";
// 1.初始化密钥
KeyPairGenerator keyPairGenerator = KeyPairGenerator.getInstance("RSA");
keyPairGenerator.initialize(51
- Hibernate学习笔记
caoyong
Hibernate
1>、Hibernate是数据访问层框架,是一个ORM(Object Relation Mapping)框架,作者为:Gavin King
2>、搭建Hibernate的开发环境
a>、添加jar包:
aa>、hibernatte开发包中/lib/required/所
- 设计模式之装饰器模式Decorator(结构型)
漂泊一剑客
Decorator
1. 概述
若你从事过面向对象开发,实现给一个类或对象增加行为,使用继承机制,这是所有面向对象语言的一个基本特性。如果已经存在的一个类缺少某些方法,或者须要给方法添加更多的功能(魅力),你也许会仅仅继承这个类来产生一个新类—这建立在额外的代码上。
- 读取磁盘文件txt,并输入String
一炮送你回车库
String
public static void main(String[] args) throws IOException {
String fileContent = readFileContent("d:/aaa.txt");
System.out.println(fileContent);
- js三级联动下拉框
3213213333332132
三级联动
//三级联动
省/直辖市<select id="province"></select>
市/省直辖<select id="city"></select>
县/区 <select id="area"></select>
- erlang之parse_transform编译选项的应用
616050468
parse_transform游戏服务器属性同步abstract_code
最近使用erlang重构了游戏服务器的所有代码,之前看过C++/lua写的服务器引擎代码,引擎实现了玩家属性自动同步给前端和增量更新玩家数据到数据库的功能,这也是现在很多游戏服务器的优化方向,在引擎层面去解决数据同步和数据持久化,数据发生变化了业务层不需要关心怎么去同步给前端。由于游戏过程中玩家每个业务中玩家数据更改的量其实是很少
- JAVA JSON的解析
darkranger
java
// {
// “Total”:“条数”,
// Code: 1,
//
// “PaymentItems”:[
// {
// “PaymentItemID”:”支款单ID”,
// “PaymentCode”:”支款单编号”,
// “PaymentTime”:”支款日期”,
// ”ContractNo”:”合同号”,
//
- POJ-1273-Drainage Ditches
aijuans
ACM_POJ
POJ-1273-Drainage Ditches
http://poj.org/problem?id=1273
基本的最大流,按LRJ的白书写的
#include<iostream>
#include<cstring>
#include<queue>
using namespace std;
#define INF 0x7fffffff
int ma
- 工作流Activiti5表的命名及含义
atongyeye
工作流Activiti
activiti5 - http://activiti.org/designer/update在线插件安装
activiti5一共23张表
Activiti的表都以ACT_开头。 第二部分是表示表的用途的两个字母标识。 用途也和服务的API对应。
ACT_RE_*: 'RE'表示repository。 这个前缀的表包含了流程定义和流程静态资源 (图片,规则,等等)。
A
- android的广播机制和广播的简单使用
百合不是茶
android广播机制广播的注册
Android广播机制简介 在Android中,有一些操作完成以后,会发送广播,比如说发出一条短信,或打出一个电话,如果某个程序接收了这个广播,就会做相应的处理。这个广播跟我们传统意义中的电台广播有些相似之处。之所以叫做广播,就是因为它只负责“说”而不管你“听不听”,也就是不管你接收方如何处理。另外,广播可以被不只一个应用程序所接收,当然也可能不被任何应
- Spring事务传播行为详解
bijian1013
javaspring事务传播行为
在service类前加上@Transactional,声明这个service所有方法需要事务管理。每一个业务方法开始时都会打开一个事务。
Spring默认情况下会对运行期例外(RunTimeException)进行事务回滚。这
- eidtplus operate
征客丶
eidtplus
开启列模式: Alt+C 鼠标选择 OR Alt+鼠标左键拖动
列模式替换或复制内容(多行):
右键-->格式-->填充所选内容-->选择相应操作
OR
Ctrl+Shift+V(复制多行数据,必须行数一致)
-------------------------------------------------------
- 【Kafka一】Kafka入门
bit1129
kafka
这篇文章来自Spark集成Kafka(http://bit1129.iteye.com/blog/2174765),这里把它单独取出来,作为Kafka的入门吧
下载Kafka
http://mirror.bit.edu.cn/apache/kafka/0.8.1.1/kafka_2.10-0.8.1.1.tgz
2.10表示Scala的版本,而0.8.1.1表示Kafka
- Spring 事务实现机制
BlueSkator
spring代理事务
Spring是以代理的方式实现对事务的管理。我们在Action中所使用的Service对象,其实是代理对象的实例,并不是我们所写的Service对象实例。既然是两个不同的对象,那为什么我们在Action中可以象使用Service对象一样的使用代理对象呢?为了说明问题,假设有个Service类叫AService,它的Spring事务代理类为AProxyService,AService实现了一个接口
- bootstrap源码学习与示例:bootstrap-dropdown(转帖)
BreakingBad
bootstrapdropdown
bootstrap-dropdown组件是个烂东西,我读后的整体感觉。
一个下拉开菜单的设计:
<ul class="nav pull-right">
<li id="fat-menu" class="dropdown">
- 读《研磨设计模式》-代码笔记-中介者模式-Mediator
bylijinnan
java设计模式
声明: 本文只为方便我个人查阅和理解,详细的分析以及源代码请移步 原作者的博客http://chjavach.iteye.com/
/*
* 中介者模式(Mediator):用一个中介对象来封装一系列的对象交互。
* 中介者使各对象不需要显式地相互引用,从而使其耦合松散,而且可以独立地改变它们之间的交互。
*
* 在我看来,Mediator模式是把多个对象(
- 常用代码记录
chenjunt3
UIExcelJ#
1、单据设置某行或某字段不能修改
//i是行号,"cash"是字段名称
getBillCardPanelWrapper().getBillCardPanel().getBillModel().setCellEditable(i, "cash", false);
//取得单据表体所有项用以上语句做循环就能设置整行了
getBillC
- 搜索引擎与工作流引擎
comsci
算法工作搜索引擎网络应用
最近在公司做和搜索有关的工作,(只是简单的应用开源工具集成到自己的产品中)工作流系统的进一步设计暂时放在一边了,偶然看到谷歌的研究员吴军写的数学之美系列中的搜索引擎与图论这篇文章中的介绍,我发现这样一个关系(仅仅是猜想)
-----搜索引擎和流程引擎的基础--都是图论,至少像在我在JWFD中引擎算法中用到的是自定义的广度优先
- oracle Health Monitor
daizj
oracleHealth Monitor
About Health Monitor
Beginning with Release 11g, Oracle Database includes a framework called Health Monitor for running diagnostic checks on the database.
About Health Monitor Checks
Health M
- JSON字符串转换为对象
dieslrae
javajson
作为前言,首先是要吐槽一下公司的脑残编译部署方式,web和core分开部署本来没什么问题,但是这丫居然不把json的包作为基础包而作为web的包,导致了core端不能使用,而且我们的core是可以当web来用的(不要在意这些细节),所以在core中处理json串就是个问题.没办法,跟编译那帮人也扯不清楚,只有自己写json的解析了.
- C语言学习八结构体,综合应用,学生管理系统
dcj3sjt126com
C语言
实现功能的代码:
# include <stdio.h>
# include <malloc.h>
struct Student
{
int age;
float score;
char name[100];
};
int main(void)
{
int len;
struct Student * pArr;
int i,
- vagrant学习笔记
dcj3sjt126com
vagrant
想了解多主机是如何定义和使用的, 所以又学习了一遍vagrant
1. vagrant virtualbox 下载安装
https://www.vagrantup.com/downloads.html
https://www.virtualbox.org/wiki/Downloads
查看安装在命令行输入vagrant
2.
- 14.性能优化-优化-软件配置优化
frank1234
软件配置性能优化
1.Tomcat线程池
修改tomcat的server.xml文件:
<Connector port="8080" protocol="HTTP/1.1" connectionTimeout="20000" redirectPort="8443" maxThreads="1200" m
- 一个不错的shell 脚本教程 入门级
HarborChung
linuxshell
一个不错的shell 脚本教程 入门级
建立一个脚本 Linux中有好多中不同的shell,但是通常我们使用bash (bourne again shell) 进行shell编程,因为bash是免费的并且很容易使用。所以在本文中笔者所提供的脚本都是使用bash(但是在大多数情况下,这些脚本同样可以在 bash的大姐,bourne shell中运行)。 如同其他语言一样
- Spring4新特性——核心容器的其他改进
jinnianshilongnian
spring动态代理spring4依赖注入
Spring4新特性——泛型限定式依赖注入
Spring4新特性——核心容器的其他改进
Spring4新特性——Web开发的增强
Spring4新特性——集成Bean Validation 1.1(JSR-349)到SpringMVC
Spring4新特性——Groovy Bean定义DSL
Spring4新特性——更好的Java泛型操作API
Spring4新
- Linux设置tomcat开机启动
liuxingguome
tomcatlinux开机自启动
执行命令sudo gedit /etc/init.d/tomcat6
然后把以下英文部分复制过去。(注意第一句#!/bin/sh如果不写,就不是一个shell文件。然后将对应的jdk和tomcat换成你自己的目录就行了。
#!/bin/bash
#
# /etc/rc.d/init.d/tomcat
# init script for tomcat precesses
- 第13章 Ajax进阶(下)
onestopweb
Ajax
index.html
<!DOCTYPE html PUBLIC "-//W3C//DTD XHTML 1.0 Transitional//EN" "http://www.w3.org/TR/xhtml1/DTD/xhtml1-transitional.dtd">
<html xmlns="http://www.w3.org/
- Troubleshooting Crystal Reports off BW
blueoxygen
BO
http://wiki.sdn.sap.com/wiki/display/BOBJ/Troubleshooting+Crystal+Reports+off+BW#TroubleshootingCrystalReportsoffBW-TracingBOE
Quite useful, especially this part:
SAP BW connectivity
For t
- Java开发熟手该当心的11个错误
tomcat_oracle
javajvm多线程单元测试
#1、不在属性文件或XML文件中外化配置属性。比如,没有把批处理使用的线程数设置成可在属性文件中配置。你的批处理程序无论在DEV环境中,还是UAT(用户验收
测试)环境中,都可以顺畅无阻地运行,但是一旦部署在PROD 上,把它作为多线程程序处理更大的数据集时,就会抛出IOException,原因可能是JDBC驱动版本不同,也可能是#2中讨论的问题。如果线程数目 可以在属性文件中配置,那么使它成为
- 正则表达式大全
yang852220741
html编程正则表达式
今天向大家分享正则表达式大全,它可以大提高你的工作效率
正则表达式也可以被当作是一门语言,当你学习一门新的编程语言的时候,他们是一个小的子语言。初看时觉得它没有任何的意义,但是很多时候,你不得不阅读一些教程,或文章来理解这些简单的描述模式。
一、校验数字的表达式
数字:^[0-9]*$
n位的数字:^\d{n}$
至少n位的数字:^\d{n,}$
m-n位的数字:^\d{m,n}$