biopython:2:序列组成
第一章:biopython
第二节:序列组成
SeqIO同时读取两个文件read1文件和read2文件,利用itertools
from Bio import SeqIO
import itertools
for r1, r2 in itertools.izip(SeqIO.parse(gzip.open('*.R1.fine.fastq.gz'),'fastq'), SeqIO.parse(gzip.open('*.R2.fine.fastq.gz'),'fastq')):
····r1,r2
1,序列的特性
生物信息当中的序列(DNA、RNA、amino acid)与python当中的字符串有两点不同(其余属性大致相同,比如序列长度,可以进行切片等)
第一点不同:序列有 translate() ,即翻译;序列有reverse_complement() ,即反向互补序列(反向互补序列一般对DNA序列而言)
第二点不同:序列存在一定的字母排列规则,即为alphabet;例如,DNA序列为“TAGC”表示四种碱基,而氨基酸序列也可以为“AGCT”表示不同的氨基酸简写,一般在文件中会显示氨基酸的一个字母的简写
2,序列属性alphabet
Bio包当中存在Bio.Alphabet模块,其作用是定义DNA、RNA及amino acid序列
amino acid序列,有 ExtendedIUPACProtein 用法,能够区分该大写字母为哪种氨基酸,例如:“U”表示“Sec”,“B”表示“Asx”,(这两种均为稀有氨基酸)而“X”表示“Xxx”表示为未知氨基酸
DNA序列,有IUPACAmbiguousDNA及ExtendedIUPACDNA用法
RNA序列,有IUPACAmbiguousRNA及IUPACUnmbiguousDNA用法
3,与序列相关的模块使用
>>> from Bio.Seq import Seq#该模块用于构建序列
>>> from Bio.Alphabet import IUPAC#该模块用于判断序列类型
>>> my_seq=Seq("AGTACA", IUPAC.unambiguous_dna)#根据Alphabet()特性将Seq定义为DNA序列
>>> my_prot=Seq("AGTACA", IUPAC.protein)#定义一个氨基酸序列
>>> my_seq.alphabet
IUPACUnambiguousDNA()
IUPAC.unambiguous_dna表示确定的DNA序列,不允许存在模糊碱基
IUPAC.ambiguous_dna表示允许存在模糊碱基
函数Seq()(首字母大写)加字符串,再加IUPAC特性,可表示DNA或氨基酸序列
构建序列方法:
Seq(str("AGTACA"), IUPAC.unambiguous_dna)
4,序列与字符串存在一些相同的特性
>>> my_seq=Seq("GATCG", IUPAC.unambiguous_dna)#定义一段DNA序列
>>> for index, letter in enumerate(my_seq):
>>> ····print "%d\t%s"%(index, letter)#index为序列编号值为0,letter为Seq
>>> len(my_seq)#序列长度
>>> my_seq[0]#序列的第一个碱基,返回一个字符串,方法类似于字符串的切片
>>> my_seq.count("A")#返回序列当中“A”碱基的个数
>>> my_seq.count("G")#计算“G”碱基个数
>>> float(my_seq.count('G')+my_seq.count('C'))/len(my_seq)*100#计算序列的GC含量
但其实在Bio.SeqUtils模块中有一个GC函数,可计算序列GC含量
>>> from Bio.SeqUtils import GC
>>> GC(my_seq)#返回一个百分数数值并不带百分号
序列的split方法和字符串的切片方式相同
>>> my_seq=Seq("GATCGATGGGCCTATA", IUPAC.unambiguous_dna)
>>> my_seq[4:12]#与字符串切片结果相同,index从0开始,不包括右边界的数值
Seq('GATGGGCC', IUPACUnambiguousDNA())
>>> my_seq.upper()
Seq('GATGGGCC', IUPACUnambiguousDNA())
>>> my_seq.lower()
Seq('gatgggcc', IUPACUnambiguousDNA())
利用字符串内置方法upper()和lower()可将DNA序列碱基进行大小写改变
5,序列与字符串的不同特性
1,反向序列及反向互补序列
>>> my_seq=Seq("GATCGATGGG", IUPAC.unambiguous_dna)
>>> my_seq.complement()#输出反向序列
Seq('CTAGCTACCC', IUPACUnambiguousDNA())
实现的结果是对应的碱基位置直接输出配对的碱基
>>> my_seq.reverse_complement()#输出反向互补序列
Seq('CCCATCGATC', IUPACUnambiguousDNA())
实现的结果是输出my_seq序列的反向互补序列;DNA序列的书写方式均为5’ -> 3’方向,所以对于DNA序列的双螺旋结构,本着碱基互补配对的原则,my_seq如果是其中的一条DNA链,而该链的反向互补序列的意义和my_seq序列是等同的,即双螺旋结构的另一条链;此时可以理解为my_seq的反向序列是3’ -> 5’方向的书写方式,但在一般的输出格式情况下是不常见的。
一般情况下只有DNA序列才能提及序列与反向互补序列的概念
2,DNA序列的转录(Transription)
>>> coding_dna=Seq("GATCGATGGG", IUPAC.unambiguous_dna)
>>> messenger_rna=coding_dna.transcribe()#转录将T改成U
>>> messenger_rna
Seq('GAUCGAUGGG', IUPACUnambiguousRNA())
>>> template_dna=coding_dna.reverse_complement()
template_dna.transcribe()
Seq('CCCAUCGAUC', IUPACUnambiguousRNA())
将DNA链转录成RNA链,用Seq中的transcribe()函数
将RNA链反转录成DNA链,用Seq中的back_transcribe()函数
>>> Seq("GAUCGAUGGG", IUPAC.unambiguous_rna).back_transcribe()
Seq('GATCGATGGG', IUPACUnambiguousDNA())
3,mRNA序列的翻译(Translation)
在实际书写时,也可以将mRNA序列书写成cDNA序列,例如:
>>> my_cdna=Seq('GATCGAATGGAACCG', IUPAC.unambiguous_dna)
>>> my_cdna.translate()
Seq('DRMEP', IUPACProtein())
如果序列当中存在终止密码子则在翻译成氨基酸序列时,终止密码子处显示的氨基酸为"*"
>>> coding_dna=Seq('ATGGCCATTGTAATGGGCCGCTGAAAGGGTGCCCGATAG', IUPAC.unambiguous_dna)
>>> coding_dna.translate()
Seq('MAIVMGR*KGAR*', HasStopCodon(IUPACProtein(), '*'))
biopython的translation tables是基于NCBI的编码规则,而biopython能够应用standard genetic code(NCBI table id 1)(coding_dna.translate() 默认是table1)
同时biopython也可以处理mitochondrial(脊椎动物线粒体序列),即为table2
>>> coding_dna.translate(table='Vertebrate Mitochondrial')
同样的书写方式也可以是coding_dna.translate(table=2)
coding_dna.translate(table=2, to_stop=True)#其中to_stop=True表示编码如果出现终止密码子不用"*"表示
coding_dna.translate(table=2, stop_symbol="@")#表示终止密码子用“@”符号代替
>>> coding_dna=Seq('ATGGCCATTGTAATGGGCCGCTGAAAGGGTGCCCGATAG', IUPAC.unambiguous_dna)
>>> coding_dna.translate()
Seq('MAIVMGR*KGAR*', HasStopCodon(IUPACProtein(), '*'))
>>> coding_dna.translate(table=2)
Seq('MAIVMGRWKGAR*', HasStopCodon(IUPACProtein(), '*'))
>>> coding_dna.translate(table=2, to_stop=True)
Seq('MAIVMGRWKGAR', IUPACProtein())
>>> coding_dna.translate(table=2, stop_symbol="@")
Seq('MAIVMGRWKGAR@', HasStopCodon(IUPACProtein(), '@'))
4,密码子表(Translation Tables)
通过以下模块可查看密码子表数量及种类
>>> from Bio.Data import CodonTable
>>> CodonTable.unambiguous_dna_by_id#通过密码子表id方法查看内容
{1:
例如,想查看其中一个密码子表可以输入:
>>> print CodonTable.unambiguous_dna_by_id[1]
Table 1 Standard, SGC0
当然也可以通过密码子表名字查看内容
>>> CodonTable.unambiguous_dna_by_name
当然也可以查看起始密码子,终止密码子,以及密码子对应的氨基酸
>>> CodonTable.unambiguous_dna_by_name['Yeast Mitochondrial'].stop_codons
['TAA', 'TAG']
>>> CodonTable.unambiguous_dna_by_name['Yeast Mitochondrial'].start_codons
['ATA', 'ATG']
>>> CodonTable.unambiguous_dna_by_name['Yeast Mitochondrial'].forward_table["ACG"]
'T'
>>> CodonTable.unambiguous_dna_by_name['Yeast Mitochondrial'].forward_table["ATA"]
'M'
>>> CodonTable.unambiguous_dna_by_name['Yeast Mitochondrial'].forward_table["ATG"]
'M'
>>> CodonTable.unambiguous_dna_by_name['Yeast Mitochondrial'].forward_table["TAC"]
'Y'
当然无法查看终止密码子的蛋白编码的氨基酸
>>> CodonTable.unambiguous_dna_by_name['Yeast Mitochondrial'].forward_table["TAG"]
Traceback (most recent call last):
··File "
KeyError: 'TAG'
>>> CodonTable.unambiguous_dna_by_name['Yeast Mitochondrial'].forward_table
{'CTT': 'T', 'ATG': 'M', 'ACA': 'T', 'ACG': 'T', 'ATC': 'I', 'ATA': 'M', 'AGG': 'R', 'CCT': 'P', 'AGC': 'S', 'AGA': 'R', 'ATT': 'I', 'CTG': 'T', 'CTA': 'T', 'ACT': 'T', 'CCG': 'P', 'AGT': 'S', 'CCA': 'P', 'CCC': 'P', 'TAT': 'Y', 'GGT': 'G', 'CGA': 'R', 'CGC': 'R', 'CGG': 'R', 'GGG': 'G', 'GGA': 'G', 'GGC': 'G', 'TAC': 'Y', 'CGT': 'R', 'GTA': 'V', 'GTC': 'V', 'GTG': 'V', 'GAG': 'E', 'GTT': 'V', 'GAC': 'D', 'GAA': 'E', 'AAG': 'K', 'AAA': 'K', 'AAC': 'N', 'CTC': 'T', 'CAT': 'H', 'AAT': 'N', 'CAC': 'H', 'CAA': 'Q', 'CAG': 'Q', 'TGT': 'C', 'TCT': 'S', 'GAT': 'D', 'TTT': 'F', 'TGC': 'C', 'TGA': 'W', 'TGG': 'W', 'TTC': 'F', 'TCG': 'S', 'TTA': 'L', 'TTG': 'L', 'TCC': 'S', 'ACC': 'T', 'TCA': 'S', 'GCA': 'A', 'GCC': 'A', 'GCG': 'A', 'GCT': 'A'}
from Bio import SeqIO
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
from Bio.Alphabet import generic_protein
from Bio.Alphabet import IUPAC
optfile=open('rename.fastq','w')
for i,read in enumerate(SeqIO.parse('test.fastq','fastq')):
····record=SeqRecord(read.seq[10:], id=str(read.seq[0:10]), description=str(i+1), letter_annotations={'phred_quality': read.letter_annotations.get('phred_quality')[10:]} )
····SeqIO.write(record,optfile,'fastq')
biopython_Tutorial.pdf page 70
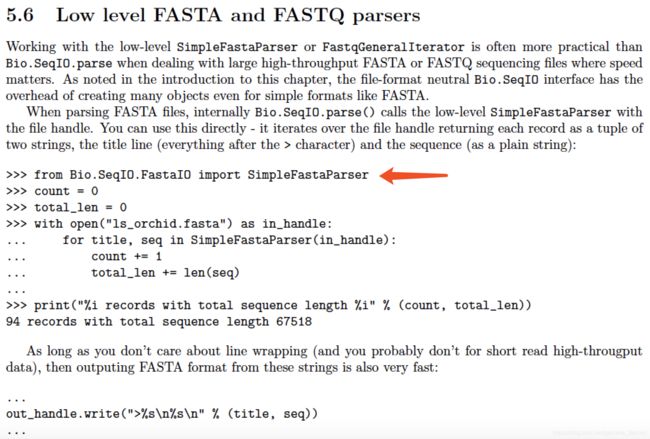
通过Bio.SeqIO.FastaIO 中的SimpleFastaParser方法,可直接将fasta格式文件读成python文件打开的形式。
for title, seq in SimpleFastaParser(in_handle)
title表示序列id,description等信息,seq表示序列字符串
out_handle.write(">%s\n%s\n" % (title, seq))
写成fasta格式文件。

同样对于fastq格式文件,利用的是Bio.SeqIO.QualityIO中的FastqGeneralIterator方法,
读取的内容是for title, seq, qual in FastqGeneralIterator(in_handle)
title表示序列id,description等信息,seq表示序列字符串,qual表示fastq文件序列的质量值。
out_handle.write("@%s\n%s\n+\n%s\n" % (title, seq, qual))
写成fastq格式文件。
将SeqRecord格式转换成fastq字符串格式或qual字符串格式
from Bio.SeqRecord import SeqRecord
对于SeqRecord格式的内容可以转换成fastq字符串格式,
>>> a=list(SeqIO.parse('test.R1.fastq','fastq'))
>>> a[0]
SeqRecord(seq=Seq(‘GGATAGAGGGGCACCACGTTCTTGCACTTCATGCTGTACAGATGCTCCATTCCT…ACG’, SingleLetterAlphabet()), id=‘NDX550197_RUO:12:HJL35AFXY:4:11511:23711:16640’, name=‘NDX550197_RUO:12:HJL35AFXY:4:11511:23711:16640’, description=‘NDX550197_RUO:12:HJL35AFXY:4:11511:23711:16640 1:N:0:CGTTAGAC+TGTCGCCA’, dbxrefs=[])
#这里a[0]是SeqIO.parse()方法读取的fastq格式文件返回的结果。
>>> a[0].format('fastq')
‘@NDX550197_RUO:12:HJL35AFXY:1:21211:25181:2255 1:N:0:CGTAAGAC+TGTCGCCA\nGGAAAGGTCTTGCACAAAGTAACATAGATGGTCCATGCCAAGGAGTTCTCCTAATTGTAAAGTTCCCCATAGGACGCAGAGAGGAGATGGGAGGTGGAGTCAGAGCTTCCTAACTACCTTATCCACATCTATATCAAGGGGCCACCCCACT\n+\nAA/AAAEAEE/AE/EEAE6EEAE66EEAEAE/E
>>> a[500].format('qual')
‘>NDX550197_RUO:12:HJL35AFXY:1:21211:25181:2255 1:N:0:CGTAAGAC+TGTCGCCA\n32 32 14 32 32 32 36 32 36 36 14 32 36 14 36 36 32 36 21 36\n36 32 36 21 21 36 36 32 36 32 36 14 36 27 32 36 32 36 36 36\n36 36 21 36 32 36 36 36 36 27 36 36 36 36 36 32 32 36 36 14\n36 14 14 21 27 27 36 36 36 36 36 21 36 36 14 36 36 36 36 21\n21 14 32 36 36 32 14 36 36 32 36 36 14 36 14 27 14 14 36 14\n32 14 36 14 36 14 14 14 14 36 14 14 36 14 36 14 14 27 14 14\n14 21 27 14 14 14 27 36 36 14 14 14 14 14 32 14 27 27 27 27\n32 32 14 14 32 32 14 32 32 32 32\n’
通过.format(‘qual’)的方法可实现将SeqRecord格式转换成qual格式,但这里得到的质量值,是字符串形式,并且除序列id外每60个字符用一个换行符分开,每一个质量值字符用空格分开。
>>> map(int, ' '.join(a[500][22:34].format('qual').split('\n')[1:-1]).split(' '))
[36, 21, 21, 36, 36, 32, 36, 32, 36, 14, 36, 27]
>>> map(int, ' '.join(a[500].format('qual').split('\n')[1:-1]).split(' '))
通过这个方法可转换成整形的列表
>>> a[500][22:34].format('fastq')
‘@NDX550197_RUO:12:HJL35AFXY:1:21211:25181:2255 1:N:0:CGTAAGAC+TGTCGCCA\nCATAGATGGTCC\n+\nE66EEAEAE/E<\n’
取一条reads的部分序列出来