- 情绪觉察日记第37天
露露_e800
今天是家庭关系规划师的第二阶最后一天,慧萍老师帮我做了个案,帮我处理了埋在心底好多年的一份恐惧,并给了我深深的力量!这几天出来学习,爸妈过来婆家帮我带小孩,妈妈出于爱帮我收拾东西,并跟我先生和婆婆产生矛盾,妈妈觉得他们没有照顾好我…。今晚回家见到妈妈,我很欣赏她并赞扬她,妈妈说今晚要跟我睡我说好,当我们俩躺在床上准备睡觉的时候,我握着妈妈的手对她说:妈妈这几天辛苦你了,你看你多利害把我们的家收拾得
- 关于沟通这件事,项目经理不需要每次都面对面进行
流程大师兄
很多项目经理都会遇到这样的问题,项目中由于事情太多,根本没有足够的时间去召开会议,那在这种情况下如何去有效地管理项目中的利益相关者?当然,不建议电子邮件也不需要开会的话,建议可以采取下面几种方式来形成有效的沟通,这几种方式可以帮助你努力的通过各种办法来保持和各方面的联系。项目经理首先要问自己几个问题,项目中哪些利益相关者是必须要进行沟通的?可以列出项目中所有的利益相关者清单,同时也整理出项目中哪些
- 机器学习与深度学习间关系与区别
ℒℴѵℯ心·动ꦿ໊ོ꫞
人工智能学习深度学习python
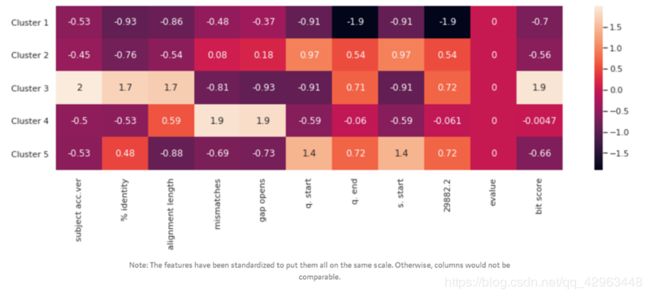
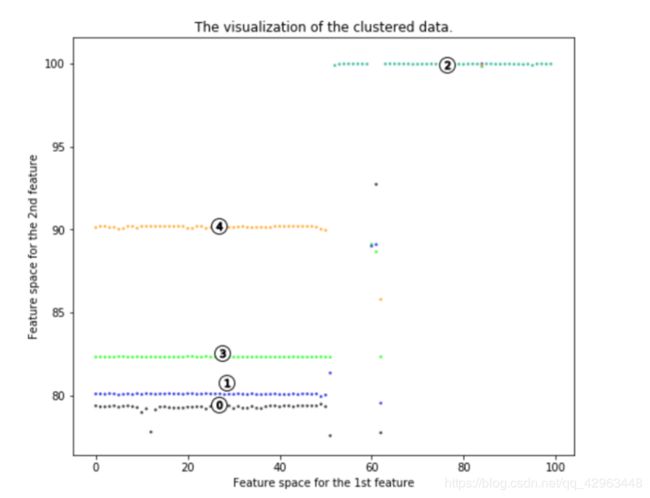
一、机器学习概述定义机器学习(MachineLearning,ML)是一种通过数据驱动的方法,利用统计学和计算算法来训练模型,使计算机能够从数据中学习并自动进行预测或决策。机器学习通过分析大量数据样本,识别其中的模式和规律,从而对新的数据进行判断。其核心在于通过训练过程,让模型不断优化和提升其预测准确性。主要类型1.监督学习(SupervisedLearning)监督学习是指在训练数据集中包含输入
- 铭刻于星(四十二)
随风至
69夜晚,绍敏同学做完功课后,看了眼房外,没听到动静才敢从书包的夹层里拿出那个心形纸团。折痕压得很深,都有些旧了,想来是已经写好很久了。绍敏同学慢慢地、轻轻地捏开折叠处,待到全部拆开后,又反复抚平纸张,然后仔细地一字字默看。只是开头的三个字是第一次看到,让她心漏跳了几拍。“亲爱的绍敏:从四年级的时候,我就喜欢你了,但是我一直不敢说,怕影响你学习。六年级的时候听说有人跟你表白,你接受了,我很难过,但
- 底层逆袭到底有多难,不甘平凡的你准备好了吗?让吴起给你说说
造命者说
底层逆袭到底有多难,不甘平凡的你准备好了吗?让吴起给你说说我叫吴起,生于公元前440年的战国初期,正是群雄并起、天下纷争不断的时候。后人说我是军事家、政治家、改革家,是兵家代表人物。评价我一生历仕鲁、魏、楚三国,通晓兵家、法家、儒家三家思想,在内政军事上都有极高的成就。周安王二十一年(公元前381年),因变法得罪守旧贵族,被人乱箭射死。我出生在卫国一个“家累万金”的富有家庭,从年轻时候起就不甘平凡
- 2020-01-25
晴岚85
郑海燕坚持分享590天2020.1.24在生活中只存在两个问题。一个问题是:你知道想要达成的目标是什么,但却不知道如何才能达成;另一个问题是:你不知道你的目标是什么。前一个是行动的问题,后一个是结果的问题。通过制定具体的下一步行动,可以解决不知道如何开始行动的问题。而通过去想象结果,对结果做预估,可以解决找不着目标的问题。对于所有吸引我们注意力,想要完成的任务,你可以先想象一下,预期的结果究竟是什
- 随笔 | 仙一般的灵气
海思沧海
仙岛今天,我看了你全部,似乎已经进入你的世界我不知道,这是否是梦幻,还是你仙一般的灵气吸引了我也许每一个人都要有一份属于自己的追求,这样才能够符合人生的梦想,生活才能够充满着阳光与快乐我不知道,我为什么会这样的感叹,是在感叹自己的人生,还是感叹自己一直没有孜孜不倦的追求只感觉虚度了光阴,每天活在自己的梦中,活在一个不真实的世界是在逃避自己,还是在逃避周围的一切有时候我嘲笑自己,嘲笑自己如此的虚无,
- 想家
爆米花机
也许不同于大家对家乡的思念,我对家乡甚至是疯狂的不舍。还未踏出车站就感觉到幸福,我享受这里的夕阳、这里的浓烈柴火味、这里每一口家常菜。我是宅女,我贪恋家的安逸。刚刚踏出大学校门,初出茅庐,无法适应每年只能国庆和春节回家。我焦虑、失眠、无端发脾气,是无法适应工作的节奏,是无法接受我将一步步离开家乡的事实。我不想承认自己胸无大志,选择再次踏上征程。图片发自App
- 【iOS】MVC设计模式
Magnetic_h
iosmvc设计模式objective-c学习ui
MVC前言如何设计一个程序的结构,这是一门专门的学问,叫做"架构模式"(architecturalpattern),属于编程的方法论。MVC模式就是架构模式的一种。它是Apple官方推荐的App开发架构,也是一般开发者最先遇到、最经典的架构。MVC各层controller层Controller/ViewController/VC(控制器)负责协调Model和View,处理大部分逻辑它将数据从Mod
- element实现动态路由+面包屑
软件技术NINI
vue案例vue.js前端
el-breadcrumb是ElementUI组件库中的一个面包屑导航组件,它用于显示当前页面的路径,帮助用户快速理解和导航到应用的各个部分。在Vue.js项目中,如果你已经安装了ElementUI,就可以很方便地使用el-breadcrumb组件。以下是一个基本的使用示例:安装ElementUI(如果你还没有安装的话):你可以通过npm或yarn来安装ElementUI。bash复制代码npmi
- 10月|愿你的青春不负梦想-读书笔记-01
Tracy的小书斋
本书的作者是俞敏洪,大家都很熟悉他了吧。俞敏洪老师是我行业的领头羊吧,也是我事业上的偶像。本日摘录他书中第一章中的金句:『一个人如果什么目标都没有,就会浑浑噩噩,感觉生命中缺少能量。能给我们能量的,是对未来的期待。第一件事,我始终为了进步而努力。与其追寻全世界的骏马,不如种植丰美的草原,到时骏马自然会来。第二件事,我始终有阶段性的目标。什么东西能给我能量?答案是对未来的期待。』读到这里的时候,我便
- 地推话术,如何应对地推过程中家长的拒绝
校师学
相信校长们在做地推的时候经常遇到这种情况:市场专员反馈家长不接单,咨询师反馈难以邀约这些家长上门,校区地推疲软,招生难。为什么?仅从地推层面分析,一方面因为家长受到的信息轰炸越来越多,对信息越来越“免疫”;而另一方面地推人员的专业能力和营销话术没有提高,无法应对家长的拒绝,对有意向的家长也不知如何跟进,眼睁睁看着家长走远;对于家长的疑问,更不知道如何有技巧地回答,机会白白流失。由于回答没技巧和专业
- C语言如何定义宏函数?
小九格物
c语言
在C语言中,宏函数是通过预处理器定义的,它在编译之前替换代码中的宏调用。宏函数可以模拟函数的行为,但它们不是真正的函数,因为它们在编译时不会进行类型检查,也不会分配存储空间。宏函数的定义通常使用#define指令,后面跟着宏的名称和参数列表,以及宏展开后的代码。宏函数的定义方式:1.基本宏函数:这是最简单的宏函数形式,它直接定义一个表达式。#defineSQUARE(x)((x)*(x))2.带参
- 微服务下功能权限与数据权限的设计与实现
nbsaas-boot
微服务java架构
在微服务架构下,系统的功能权限和数据权限控制显得尤为重要。随着系统规模的扩大和微服务数量的增加,如何保证不同用户和服务之间的访问权限准确、细粒度地控制,成为设计安全策略的关键。本文将讨论如何在微服务体系中设计和实现功能权限与数据权限控制。1.功能权限与数据权限的定义功能权限:指用户或系统角色对特定功能的访问权限。通常是某个用户角色能否执行某个操作,比如查看订单、创建订单、修改用户资料等。数据权限:
- 理解Gunicorn:Python WSGI服务器的基石
范范0825
ipythonlinux运维
理解Gunicorn:PythonWSGI服务器的基石介绍Gunicorn,全称GreenUnicorn,是一个为PythonWSGI(WebServerGatewayInterface)应用设计的高效、轻量级HTTP服务器。作为PythonWeb应用部署的常用工具,Gunicorn以其高性能和易用性著称。本文将介绍Gunicorn的基本概念、安装和配置,帮助初学者快速上手。1.什么是Gunico
- 《投行人生》读书笔记
小蘑菇的树洞
《投行人生》----作者詹姆斯-A-朗德摩根斯坦利副主席40年的职业洞见-很短小精悍的篇幅,比较适合初入职场的新人。第一部分成功的职业生涯需要规划1.情商归为适应能力分享与协作同理心适应能力,更多的是自我意识,你有能力识别自己的情并分辨这些情绪如何影响你的思想和行为。2.对于初入职场的人的建议,细节,截止日期和数据很重要截止日期,一种有效的方法是请老板为你所有的任务进行优先级排序。和老板喝咖啡的好
- swagger访问路径
igotyback
swagger
Swagger2.x版本访问地址:http://{ip}:{port}/{context-path}/swagger-ui.html{ip}是你的服务器IP地址。{port}是你的应用服务端口,通常为8080。{context-path}是你的应用上下文路径,如果应用部署在根路径下,则为空。Swagger3.x版本对于Swagger3.x版本(也称为OpenAPI3)访问地址:http://{ip
- mysql禁用远程登录
igotyback
mysql
去mysql库中的user表里,将host都改成localhost之后刷新权限FLUSHPRIVILEGES;
- 如何在 Fork 的 GitHub 项目中保留自己的修改并同步上游更新?github_fork_update
iBaoxing
github
如何在Fork的GitHub项目中保留自己的修改并同步上游更新?在GitHub上Fork了一个项目后,你可能会对项目进行一些修改,同时原作者也在不断更新。如果想要在保留自己修改的基础上,同步原作者的最新更新,很多人会不知所措。本文将详细讲解如何在不丢失自己改动的情况下,将上游仓库的更新合并到自己的仓库中。问题描述假设你在GitHub上Fork了一个项目,并基于该项目做了一些修改,随后你发现原作者对
- 扫地机类清洁产品之直流无刷电机控制
悟空胆好小
清洁服务机器人单片机人工智能
扫地机类清洁产品之直流无刷电机控制1.1前言扫地机产品有很多的电机控制,滚刷电机1个,边刷电机1-2个,清水泵电机,风机一个,部分中高端产品支持抹布功能,也就是存在抹布盘电机,还有追觅科沃斯石头等边刷抬升电机,滚刷抬升电机等的,这些电机有直流有刷电机,直接无刷电机,步进电机,电磁阀,挪动泵等不同类型。电机的原理,驱动控制方式也不行。接下来一段时间的几个文章会作个专题分析分享。直流有刷电机会自动持续
- 店群合一模式下的社区团购新发展——结合链动 2+1 模式、AI 智能名片与 S2B2C 商城小程序源码
说私域
人工智能小程序
摘要:本文探讨了店群合一的社区团购平台在当今商业环境中的重要性和优势。通过分析店群合一模式如何将互联网社群与线下终端紧密结合,阐述了链动2+1模式、AI智能名片和S2B2C商城小程序源码在这一模式中的应用价值。这些创新元素的结合为社区团购带来了新的机遇,提升了用户信任感、拓展了营销渠道,并实现了线上线下的完美融合。一、引言随着互联网技术的不断发展,社区团购作为一种新兴的商业模式,在满足消费者日常需
- 2021-08-26
影幽
在生活中,女人与男人的感悟往往有所不同。人生最大的舞台就是生活,大幕随时都可能拉开,关键是你愿不愿意表演都无法躲避。在生活中,遇事不要急躁,不要急于下结论,尤其生气时不要做决断,要学会换位思考,大事化小小事化了,把复杂的事情尽量简单处理,千万不要把简单的事情复杂化。永远不要扭曲,别人善意,无药可救。昨天是张过期的支票,明天是张信用卡,只有今天才是现金,要善加利用!执着的攀登者不必去与别人比较自己的
- html 中如何使用 uniapp 的部分方法
某公司摸鱼前端
htmluni-app前端
示例代码:Documentconsole.log(window);效果展示:好了,现在就可以uni.使用相关的方法了
- ArcGIS栅格计算器常见公式(赋值、0和空值的转换、补充栅格空值)
研学随笔
arcgis经验分享
我们在使用ArcGIS时通常经常用到栅格计算器,今天主要给大家介绍我日常中经常用到的几个公式,供大家参考学习。将特定值(-9999)赋值为0,例如-9999.Con("raster"==-9999,0,"raster")2.给空值赋予特定的值(如0)Con(IsNull("raster"),0,"raster")3.将特定的栅格值(如1)赋值为空值,其他保留原值SetNull("raster"==
- 高级编程--XML+socket练习题
masa010
java开发语言
1.北京华北2114.8万人上海华东2,500万人广州华南1292.68万人成都华西1417万人(1)使用dom4j将信息存入xml中(2)读取信息,并打印控制台(3)添加一个city节点与子节点(4)使用socketTCP协议编写服务端与客户端,客户端输入城市ID,服务器响应相应城市信息(5)使用socketTCP协议编写服务端与客户端,客户端要求用户输入city对象,服务端接收并使用dom4j
- 抖音乐买买怎么加入赚钱?赚钱方法是什么
测评君高省
你会在抖音买东西吗?如果会,那么一定要免费注册一个乐买买,抖音直播间,橱窗,小视频里的小黄车买东西都可以返佣金!省下来都是自己的,分享还可以赚钱乐买买是好省旗下的抖音返佣平台,乐买买分析社交电商的价值,乐买买属于今年难得的副业项目风口机会,2019年错过做好省的搞钱的黄金时期,那么2022年千万别再错过乐买买至于我为何转到高省呢?当然是高省APP佣金更高,模式更好,终端用户不流失。【高省】是一个自
- 2018-07-23-催眠日作业-#不一样的31天#-66小鹿
小鹿_33
预言日:人总是在逃避命运的路上,与之不期而遇。心理学上有个著名的名词,叫做自证预言;经济学上也有一个很著名的定律叫做,墨菲定律;在灵修派上,还有一个很著名的法则,叫做吸引力法则。这3个领域的词,虽然看起来不太一样,但是他们都在告诉人们一个现象:你越担心什么,就越有可能会发生什么。同样的道理,你越想得到什么,就应该要积极地去创造什么。无论是自证预言,墨菲定律还是吸引力法则,对人都有正反2个维度的影响
- 我的烦恼
余建梅
我的烦恼。女儿问我:“你给学生布置什么作文题目?”“《我的烦恼》。”“他们都这么大了,你觉得他们还有烦恼吗?”“有啊!每个人都会有自己烦恼。”“我不相信,大人是没有烦恼的,如果说一定有的话,你的烦恼和我写作业有关,而且是小烦恼。不像我,天天被你说,有这样的妈妈,烦恼是没完没了。”女儿愤愤不平。每个人都会有自己的烦恼,处在上有老下有小的年纪,烦恼多的数不完。想干好工作带好孩子,想孝顺父母又想经营好自
- 【一起学Rust | 设计模式】习惯语法——使用借用类型作为参数、格式化拼接字符串、构造函数
广龙宇
一起学Rust#Rust设计模式rust设计模式开发语言
提示:文章写完后,目录可以自动生成,如何生成可参考右边的帮助文档文章目录前言一、使用借用类型作为参数二、格式化拼接字符串三、使用构造函数总结前言Rust不是传统的面向对象编程语言,它的所有特性,使其独一无二。因此,学习特定于Rust的设计模式是必要的。本系列文章为作者学习《Rust设计模式》的学习笔记以及自己的见解。因此,本系列文章的结构也与此书的结构相同(后续可能会调成结构),基本上分为三个部分
- 放下是一段成长的修行
小莳玥
人来到这个世界上,只有两件事:生和死。一件事已经做完了,另一件你还急什么呢?是人,都有七情六欲。是心,都有喜怒哀乐,这些再正常不过了。别总抱怨自己活得累,过得辛苦。永远记住:舒坦是留给死人的。苦,才是生活;累,才是工作;变,才是命运;忍,才是历练;容,才是智慧;静,才是修养;舍,才会得到;做,才会拥有。人生,活得太清楚,才是最大的不明白。有些事,看得很清,却说不清;有些人,了解很深,却猜不透;有些
- [黑洞与暗粒子]没有光的世界
comsci
无论是相对论还是其它现代物理学,都显然有个缺陷,那就是必须有光才能够计算
但是,我相信,在我们的世界和宇宙平面中,肯定存在没有光的世界....
那么,在没有光的世界,光子和其它粒子的规律无法被应用和考察,那么以光速为核心的
&nbs
- jQuery Lazy Load 图片延迟加载
aijuans
jquery
基于 jQuery 的图片延迟加载插件,在用户滚动页面到图片之后才进行加载。
对于有较多的图片的网页,使用图片延迟加载,能有效的提高页面加载速度。
版本:
jQuery v1.4.4+
jQuery Lazy Load v1.7.2
注意事项:
需要真正实现图片延迟加载,必须将真实图片地址写在 data-original 属性中。若 src
- 使用Jodd的优点
Kai_Ge
jodd
1. 简化和统一 controller ,抛弃 extends SimpleFormController ,统一使用 implements Controller 的方式。
2. 简化 JSP 页面的 bind, 不需要一个字段一个字段的绑定。
3. 对 bean 没有任何要求,可以使用任意的 bean 做为 formBean。
使用方法简介
- jpa Query转hibernate Query
120153216
Hibernate
public List<Map> getMapList(String hql,
Map map) {
org.hibernate.Query jpaQuery = entityManager.createQuery(hql);
if (null != map) {
for (String parameter : map.keySet()) {
jp
- Django_Python3添加MySQL/MariaDB支持
2002wmj
mariaDB
现状
首先,
[email protected] 中默认的引擎为 django.db.backends.mysql 。但是在Python3中如果这样写的话,会发现 django.db.backends.mysql 依赖 MySQLdb[5] ,而 MySQLdb 又不兼容 Python3 于是要找一种新的方式来继续使用MySQL。 MySQL官方的方案
首先据MySQL文档[3]说,自从MySQL
- 在SQLSERVER中查找消耗IO最多的SQL
357029540
SQL Server
返回做IO数目最多的50条语句以及它们的执行计划。
select top 50
(total_logical_reads/execution_count) as avg_logical_reads,
(total_logical_writes/execution_count) as avg_logical_writes,
(tot
- spring UnChecked 异常 官方定义!
7454103
spring
如果你接触过spring的 事物管理!那么你必须明白 spring的 非捕获异常! 即 unchecked 异常! 因为 spring 默认这类异常事物自动回滚!!
public static boolean isCheckedException(Throwable ex)
{
return !(ex instanceof RuntimeExcep
- mongoDB 入门指南、示例
adminjun
javamongodb操作
一、准备工作
1、 下载mongoDB
下载地址:http://www.mongodb.org/downloads
选择合适你的版本
相关文档:http://www.mongodb.org/display/DOCS/Tutorial
2、 安装mongoDB
A、 不解压模式:
将下载下来的mongoDB-xxx.zip打开,找到bin目录,运行mongod.exe就可以启动服务,默
- CUDA 5 Release Candidate Now Available
aijuans
CUDA
The CUDA 5 Release Candidate is now available at http://developer.nvidia.com/<wbr></wbr>cuda/cuda-pre-production. Now applicable to a broader set of algorithms, CUDA 5 has advanced fe
- Essential Studio for WinRT网格控件测评
Axiba
JavaScripthtml5
Essential Studio for WinRT界面控件包含了商业平板应用程序开发中所需的所有控件,如市场上运行速度最快的grid 和chart、地图、RDL报表查看器、丰富的文本查看器及图表等等。同时,该控件还包含了一组独特的库,用于从WinRT应用程序中生成Excel、Word以及PDF格式的文件。此文将对其另外一个强大的控件——网格控件进行专门的测评详述。
网格控件功能
1、
- java 获取windows系统安装的证书或证书链
bewithme
windows
有时需要获取windows系统安装的证书或证书链,比如说你要通过证书来创建java的密钥库 。
有关证书链的解释可以查看此处 。
public static void main(String[] args) {
SunMSCAPI providerMSCAPI = new SunMSCAPI();
S
- NoSQL数据库之Redis数据库管理(set类型和zset类型)
bijian1013
redis数据库NoSQL
4.sets类型
Set是集合,它是string类型的无序集合。set是通过hash table实现的,添加、删除和查找的复杂度都是O(1)。对集合我们可以取并集、交集、差集。通过这些操作我们可以实现sns中的好友推荐和blog的tag功能。
sadd:向名称为key的set中添加元
- 异常捕获何时用Exception,何时用Throwable
bingyingao
用Exception的情况
try {
//可能发生空指针、数组溢出等异常
} catch (Exception e) {
- 【Kafka四】Kakfa伪分布式安装
bit1129
kafka
在http://bit1129.iteye.com/blog/2174791一文中,实现了单Kafka服务器的安装,在Kafka中,每个Kafka服务器称为一个broker。本文简单介绍下,在单机环境下Kafka的伪分布式安装和测试验证 1. 安装步骤
Kafka伪分布式安装的思路跟Zookeeper的伪分布式安装思路完全一样,不过比Zookeeper稍微简单些(不
- Project Euler
bookjovi
haskell
Project Euler是个数学问题求解网站,网站设计的很有意思,有很多problem,在未提交正确答案前不能查看problem的overview,也不能查看关于problem的discussion thread,只能看到现在problem已经被多少人解决了,人数越多往往代表问题越容易。
看看problem 1吧:
Add all the natural num
- Java-Collections Framework学习与总结-ArrayDeque
BrokenDreams
Collections
表、栈和队列是三种基本的数据结构,前面总结的ArrayList和LinkedList可以作为任意一种数据结构来使用,当然由于实现方式的不同,操作的效率也会不同。
这篇要看一下java.util.ArrayDeque。从命名上看
- 读《研磨设计模式》-代码笔记-装饰模式-Decorator
bylijinnan
java设计模式
声明: 本文只为方便我个人查阅和理解,详细的分析以及源代码请移步 原作者的博客http://chjavach.iteye.com/
import java.io.BufferedOutputStream;
import java.io.DataOutputStream;
import java.io.FileOutputStream;
import java.io.Fi
- Maven学习(一)
chenyu19891124
Maven私服
学习一门技术和工具总得花费一段时间,5月底6月初自己学习了一些工具,maven+Hudson+nexus的搭建,对于maven以前只是听说,顺便再自己的电脑上搭建了一个maven环境,但是完全不了解maven这一强大的构建工具,还有ant也是一个构建工具,但ant就没有maven那么的简单方便,其实简单点说maven是一个运用命令行就能完成构建,测试,打包,发布一系列功
- [原创]JWFD工作流引擎设计----节点匹配搜索算法(用于初步解决条件异步汇聚问题) 补充
comsci
算法工作PHP搜索引擎嵌入式
本文主要介绍在JWFD工作流引擎设计中遇到的一个实际问题的解决方案,请参考我的博文"带条件选择的并行汇聚路由问题"中图例A2描述的情况(http://comsci.iteye.com/blog/339756),我现在把我对图例A2的一个解决方案公布出来,请大家多指点
节点匹配搜索算法(用于解决标准对称流程图条件汇聚点运行控制参数的算法)
需要解决的问题:已知分支
- Linux中用shell获取昨天、明天或多天前的日期
daizj
linuxshell上几年昨天获取上几个月
在Linux中可以通过date命令获取昨天、明天、上个月、下个月、上一年和下一年
# 获取昨天
date -d 'yesterday' # 或 date -d 'last day'
# 获取明天
date -d 'tomorrow' # 或 date -d 'next day'
# 获取上个月
date -d 'last month'
#
- 我所理解的云计算
dongwei_6688
云计算
在刚开始接触到一个概念时,人们往往都会去探寻这个概念的含义,以达到对其有一个感性的认知,在Wikipedia上关于“云计算”是这么定义的,它说:
Cloud computing is a phrase used to describe a variety of computing co
- YII CMenu配置
dcj3sjt126com
yii
Adding id and class names to CMenu
We use the id and htmlOptions to accomplish this. Watch.
//in your view
$this->widget('zii.widgets.CMenu', array(
'id'=>'myMenu',
'items'=>$this-&g
- 设计模式之静态代理与动态代理
come_for_dream
设计模式
静态代理与动态代理
代理模式是java开发中用到的相对比较多的设计模式,其中的思想就是主业务和相关业务分离。所谓的代理设计就是指由一个代理主题来操作真实主题,真实主题执行具体的业务操作,而代理主题负责其他相关业务的处理。比如我们在进行删除操作的时候需要检验一下用户是否登陆,我们可以删除看成主业务,而把检验用户是否登陆看成其相关业务
- 【转】理解Javascript 系列
gcc2ge
JavaScript
理解Javascript_13_执行模型详解
摘要: 在《理解Javascript_12_执行模型浅析》一文中,我们初步的了解了执行上下文与作用域的概念,那么这一篇将深入分析执行上下文的构建过程,了解执行上下文、函数对象、作用域三者之间的关系。函数执行环境简单的代码:当调用say方法时,第一步是创建其执行环境,在创建执行环境的过程中,会按照定义的先后顺序完成一系列操作:1.首先会创建一个
- Subsets II
hcx2013
set
Given a collection of integers that might contain duplicates, nums, return all possible subsets.
Note:
Elements in a subset must be in non-descending order.
The solution set must not conta
- Spring4.1新特性——Spring缓存框架增强
jinnianshilongnian
spring4
目录
Spring4.1新特性——综述
Spring4.1新特性——Spring核心部分及其他
Spring4.1新特性——Spring缓存框架增强
Spring4.1新特性——异步调用和事件机制的异常处理
Spring4.1新特性——数据库集成测试脚本初始化
Spring4.1新特性——Spring MVC增强
Spring4.1新特性——页面自动化测试框架Spring MVC T
- shell嵌套expect执行命令
liyonghui160com
一直都想把expect的操作写到bash脚本里,这样就不用我再写两个脚本来执行了,搞了一下午终于有点小成就,给大家看看吧.
系统:centos 5.x
1.先安装expect
yum -y install expect
2.脚本内容:
cat auto_svn.sh
#!/bin/bash
- Linux实用命令整理
pda158
linux
0. 基本命令 linux 基本命令整理
1. 压缩 解压 tar -zcvf a.tar.gz a #把a压缩成a.tar.gz tar -zxvf a.tar.gz #把a.tar.gz解压成a
2. vim小结 2.1 vim替换 :m,ns/word_1/word_2/gc
- 独立开发人员通向成功的29个小贴士
shoothao
独立开发
概述:本文收集了关于独立开发人员通向成功需要注意的一些东西,对于具体的每个贴士的注解有兴趣的朋友可以查看下面标注的原文地址。
明白你从事独立开发的原因和目的。
保持坚持制定计划的好习惯。
万事开头难,第一份订单是关键。
培养多元化业务技能。
提供卓越的服务和品质。
谨小慎微。
营销是必备技能。
学会组织,有条理的工作才是最有效率的。
“独立
- JAVA中堆栈和内存分配原理
uule
java
1、栈、堆
1.寄存器:最快的存储区, 由编译器根据需求进行分配,我们在程序中无法控制.2. 栈:存放基本类型的变量数据和对象的引用,但对象本身不存放在栈中,而是存放在堆(new 出来的对象)或者常量池中(字符串常量对象存放在常量池中。)3. 堆:存放所有new出来的对象。4. 静态域:存放静态成员(static定义的)5. 常量池:存放字符串常量和基本类型常量(public static f