序列比较(上篇)
认识序列
蛋白质序列
由20个不同的字母(氨基酸)排列组合而成。
核酸序列
包括DNA序列和RNA序列。由4个不同的字母(碱基)排列组合而成。
FASTA格式
第一行:大于号加名称或其它注释。
第二行以后:每行60个字母(也有80的,不一定)。
序列相似性
数据库中的序列相似性搜索
对于一个蛋白质或核酸序列,你需要从序列数据库中找到与它相同或相似的序列。不可能再用眼睛去比较每一对序列,因为数据库中有太多序列,甚至用眼睛去比较一对序列都是不可能做到的。
序列相似性的重要性
相似的序列往往起源于一个共同的祖先序列。它们很可能有相似的空间结构和生物学功能,因此对于一个已知序列但未知结构和功能的蛋白质,如果与它序列相似的某些蛋白质的结构和功能已知,则可以推测这个未知结构和功能的蛋白质的结构和功能。
序列一致度(identity)与相似度(similarity)
一致度:如果两个序列(蛋白质或核酸)长度相同,那么它们的一致度定义为他们对应位置上相同的残基(一个字母,氨基酸或碱基)的数目占总长度的百分数。
相似度:如果两个序列(蛋白质或核酸)长度相同,那么它们的相似度定义为他们对应位置上相似的残基与相同的残基的数目和占总长度的百分数。
替换记分矩阵
替换记分矩阵(Substitution Matrix):反映残基之间相互替换率的矩阵,它描述了残基两两相似的量化关系。分为DNA替换记分矩阵和蛋白质替换记分矩阵。矩阵中元素值大于零,则两个残基相似;否则两个残基不相似。
核酸序列的替换记分矩阵
等价矩阵(unitary matrix)
最简单的替换记分矩阵,其中,相同核苷酸之间的匹配得分为1,不同核苷酸间的替换得分为0。由于不含有碱基的理化信息和不区别对待不同的替换,在实际的序列比较中使用较少。
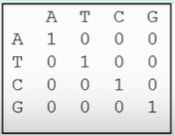 图 1 等价矩阵
图 1 等价矩阵
转换-颠换矩阵(transition-transversion matrix)
核酸的碱基按照环结构特征被划分为两类,一类是嘌呤(腺嘌呤A、鸟嘌呤G),它们有两个环;另一类是嘧啶(胞嘧啶C、胸腺嘧啶T),它们只有一个环。如果DNA碱基的替换保持环数不变,则称为转换,即嘌呤变嘌呤、嘧啶变嘧啶;如果环数发生变化,则称为颠换,即嘌呤变嘧啶、嘧啶变嘌呤。在进化过程中,转换发生的频率远比颠换高。为了反映这一情况,通常该矩阵中转换得分为-1,而颠换的得分为-5。
 图 2 转换-颠换矩阵
图 2 转换-颠换矩阵
BLAST矩阵
经过大量实际比对发现,如果令被比对的两个核苷酸相同时得分为+5,反之为-4,则比对效果较好。这个矩阵广泛被DNA序列比较所采用。
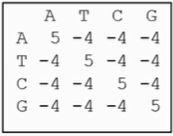 图 3 BLAST矩阵
图 3 BLAST矩阵
蛋白质的替换记分矩阵
等价矩阵(unitary matrix)
与DNA等价矩阵道理相同,相同氨基酸之间的匹配得分为1,不同氨基酸间的替换得分为0。在实际的序列比对中较少使用。
PAM矩阵(Dayhoff突变数据矩阵)
PAM矩阵基于进化原理。如果两种氨基酸替换频繁,说明自然界易接受这种替换,那么这对氨基酸替换得分就应该高。PAM矩阵是目前蛋白质序列比较中最广泛使用的记分方法之一,基础的PAM-1矩阵反应的是进化产生的每一百个氨基酸平均发生一个突变的量值。PAM-1自乘n次,可以得到PAM-n,即发生了更多次突变。
BLOSUM矩阵(blocks substitution matrix)
BLOSUM矩阵是通过关系较远的序列来获得矩阵元素的。PAM-1矩阵的产生是基于相似度较高(>85%)的序列比对,那些进化距离较远的矩阵,如PAM-250,是通过PAM-1自乘得到的。即,BLOSUM矩阵的相似度是根据真实数据产生的,而PAM矩阵是通过自乘外推而来的。和PAM矩阵一样,BLOSUM矩阵也有不同的编号,如BLOSUM-80,BLOSUM-62。80代表该矩阵是由一致度>80%的序列计算而来,同理,62指该矩阵由一致度>62%的序列计算而来。
| 氨基酸差异% |
PAM |
BLOSUM |
| 1 |
PAM-1 |
BLOSUM-99 |
| 10 |
PAM-11 |
BLOSUM-90 |
| 20 |
PAM-23 |
BLOSUM-80 |
| 30 |
PAM-38 |
BLOSUM-70 |
| 40 |
PAM-56 |
BLOSUM-60 |
| 50 |
PAM-80 |
BLOSUM-50 |
| 60 |
PAM-112 |
BLOSUM-40 |
| 70 |
PAM-159 |
BLOSUM-30 |
| 80 |
PAM-246 |
BLOSUM-20 |
总结:对于关系较远的序列的比较,由于PAM-250是推算而来,所以其准确度受到一定限制,BLOSUM-45更具优势。对于关系较近的序列之间的比较,用PAM或BLOSUM矩阵做出的比对结果,差别不大。
最常用的:BLOSUM-62
 图 4 PAM与BLOSUM的选择
图 4 PAM与BLOSUM的选择
遗传密码矩阵(genetic code matrix GCM)
遗传密码矩阵通过计算一个氨基酸转换成另一个氨基酸所需的密码子变化的数目而得到,矩阵的值对应为据此付出的代价。如果变化一个碱基就可以使一个氨基酸的密码子转换为另一个氨基酸的密码子,则这两个氨基酸的替换代价为1;如果需要2个碱基的改变,则替换代价为2,再比如从Met到Tyr三个密码子都需要变,则代价为3。
遗传密码矩阵常用于进化距离的计算,其优点是计算结果可以直接用于绘制进化树,但是它在蛋白质序列比对(尤其是相似程度很低的蛋白质序列比对)中,很少被使用。
 图 5 遗传密码矩阵
图 5 遗传密码矩阵
疏水矩阵
根据氨基酸残基替换前后疏水性的变化而得到得分矩阵。若一次氨基酸替换疏水性特性不发生太大变化,则这种替换得分高,否则替换得分低。该矩阵物理意义明确(亲水性与疏水性),有一定的理化性质依据,适用于偏重蛋白质功能方面的序列比对。
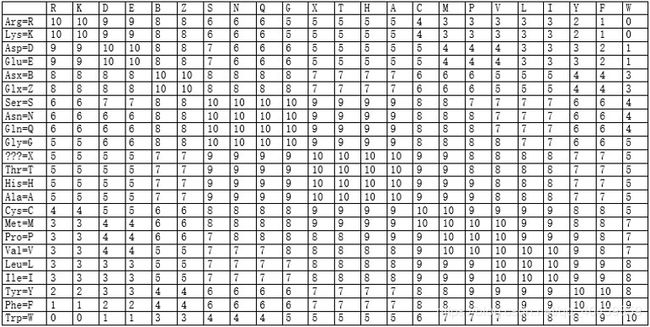 图 6 疏水矩阵
图 6 疏水矩阵
比较两个序列的方法之打点法
打点法是最简单的比较两个序列的方法,理论上可以用纸和笔完成。
假设待比较序列为Seq1:THEFASTCAT,Seq2:THEFATCAT
 图 7 打点法实例
图 7 打点法实例
依次横横竖竖比较每一个位置上的残基,相同则在这个位置上打一个点,不同则什么也不干。
结论:连续的对角线及对角线的平行线代表两条序列中相同的区域。
可以用一条序列自己对自己打点,从而可以发现序列中重复的片段。这样的打点矩阵必然是对称的,并且有一条主对角线。在横向或纵向上,与主对角线平行的短平行线所对象的片段就是重复的部分。
 图 8 序列自己和自己打点寻找重复序列
图 8 序列自己和自己打点寻找重复序列
打点法在线软件:Dotlet
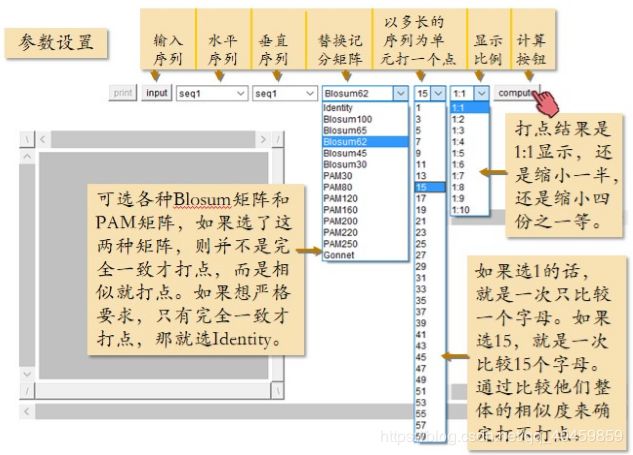 图 9 Dotlet参数设置
图 9 Dotlet参数设置
比较两个序列的方法之序列比对法
序列比对(alignment)是运用特定的算法找出两个或多个序列之间产生最大相似度得分的空格插入和序列排列方案。通俗来讲,就是通过插入空位,让上下两行中尽可能多的一致的和相似的字符对在一起。
序列s:LQRHKRTHTGEKPYE-CNQCGKAFAQ-
序列t:LQRHKRTHTGEKPYMNVINMVKPLHNS
双序列全局比对
全局比对(global alignment):用于比较两个长度近似的序列。
Needleman-Wunsch算法。1970年,SaulNeedleman和Christian Wunsch两人首先将动态规划算法应用于两条序列的全局比对,这个算法后称为Needleman-Wunsch算法。
双序列局部比对
局部比对(local alignment):用于比较一长一短两条序列。
1981年Temple Smith和Michael Waterman对局部比对进行了研究,产生了Smith-Waterman算法。
一致性和相似性
一致性(identity)= 一致字符的个数/全局比对长度
相似度(similarity)= 一致及相似的字符的个数/全局比对长度
无论两个序列长度是否相同,都要先做双序列全局比对,然后根据比对结果及比对长度计算它们的一致度和相似度。