扩增子、宏基因组测序问题集锦

扩增子、宏基因组测序问题集锦
原文转自诺禾致源,点我阅读原文。作者整理的非常好,值得学习。但本人又结合自己经验进行了修改,并对每个问题例举了实例和添加自己的理解(个人经验部分)。
微生物,是地球上最古老的生命形式之一,它们虽然微小,却无处不在。随着高通量测序技术的发展,测序成本逐步降低,测序通量飞速提高,如今我们可以用更低的成本,对微生物进行更深入和更广泛的研究。在微生物群落多样性研究中,目前主要的技术包括扩增子测序和宏基因组测序。今天杨萍给大家总结了10个扩增子和宏基因组测序中常见的问题,希望其中恰好也有您想要问的问题哦~
扩增子常见问题
01 实验室检测的DNA浓度很高,送到公司检测之后浓度却比较低呢?
- 老师在实验室多采用Nanodrop对DNA浓度进行检测,而在公司我们会结合Qubit、Nanodrop、琼脂糖电泳三种方法检测DNA样品的质量;
- 由于不同检测方法的原理不同,所以检测出的结果也会存在一定的差异。其中,Nanodrop检测法是基于紫外分光光度原理进行检测,由于DNA样品中可能含有部分杂质,因此会造成结果虚高的现象;Qubit检测法则是基于荧光标记的原理进行检测,结果会更准确;
- 当两种检测方法的结果出现差异时,我们以Qubit检测结果为准。
个人经验:我用CTAB法提取的小麦总DNA, Nanodrop检测浓度大于1000 ng/ul,结果公司返回的检测报告只有100 ng/ul,差别可达10倍。可能是植物多糖含量高,DNA纯度比较难保证。
02 在计算微生物群落样品之间的距离时,分别基于加权与非加权两种不同的算法绘制出的结果展示图有什么不同?如何进行选择呢?
- 在计算微生物群落样品之间的距离时,加权是考虑到样品中OTUs的相对丰度信息,而非加权则没有考虑物种的相对丰度信息;
- 如果老师研究的生物学问题与物种的相对丰度信息密切相关,使用加权算法的结果展示可能更为符合;如果研究的生物问题与丰度关系不密切,或者各组的区分与低丰度的OTUs更为密切,则使用非加权的结果可能更为合适。
个人经验:我们组研究的一般基因型等差别对微生物组的影响,权重是非常重要的,非加权(unweighted Unifrac)的结果乱成一团,完全不适合;即使是加权的(weight unifrac)解释也不好,感觉它们比较适合区分差别较大的不同生态位(niche)。我们用bray-curtis物种距离一般会有更好的解释。
03 在韦恩图中,为什么组中OTU个数与单个样本个数的加和不一致?
对于组的OTU计数,采用的是取并集的方式(当该组的重复样品中只要有一个样品存在该OTU,那么就认为该组内存在该OTU,若所有重复样品中都不存在该OTU,即认为该组内不存在该OTU)。
个人经验:样品和组间共有、特有OTU的结果很不可信,因为OTU的数量受测序深度和随机因素影响很大。其次,在高通量测序的结果中,大数据中出现0或1、2、3在统计上并没有显著差异,更多是随机分布的假阳性。建议关注差异OTU的类别,不要在此处不准确的结果上浪费时间。
04 如何选择T-test、 Metastat及LEFSe的结果?
由于这三种统计分析方法所使用的统计检验的方法有所不同,因此得出的结果也会存在差异。其中,T-test使用的是t检验的方法,Metastat会根据样本情况自动调整统计的方法(秩和检验或fisher检验),而LEfSe则使用了秩和检验和线性判别分析(LDA),这3种统计分析方法筛选结果均是可信的,老师可以根据自己的研究背景选择最为符合的分析结果。
05 对于生物学重复偏离较大的样本,如何进行分析?
生物学重复通常建议5个以上,至少3个。对于重复样品间存在较大差异的个别样本,一般建议:
1. 从样品的准备过程进行分析,生物学重复的样品,除了和设定的分组条件有关外,可能还受到很多其他因素的影响,进而造成分析结果出现差异;
2. 对于出现显著离群的个别样本,推测可能为样本自身的原因(如在采样、保藏、提取、扩增过程中样本出现了问题等),建议剔除该样本后,再进行分析。
个人经验:偏离较大的个别样品,对整体的统计是影响不大的,如果不是明显人为原因的错误,不建议原始数据随便删除此样品。如果出现多个样品出现异常,比如分为差别很大的两类,要检查操作中是与有影响的步骤,如种子混杂,分批取材、提取和扩增是否使用不同方法或试剂、barcode或index是否有偏好,建库和测序是否同批等,找不到原因可再完全重复实验验证,确保实验结果准确是最重要的。
宏基因组常见问题
01 在组装过程中,组装后的基因为什么不完整?
宏基因组组装的效果主要跟以下几个因素有关:样本的测序数据量,物种的多样性,物种丰度分布不均匀等,这些因素都会造成宏基因组组装比细菌等单物种的组装更加困难,这也是目前宏基因组研究中有待突破的重点。
02 16S扩增子和宏基因组分析结果存在差别的原因?
- 两者的分析方法存在较大差异:16S是先扩增后测序,而且不同物种DNA的扩增倍数也不一致;在宏基因组DNA测序中,测序深度可能不是十分充分,并且宏基因组分析得到的相对物种丰度的差异与DNA提取以及测序的方法都密切相关;
- 两者采用的物种注释方法及数据库都存在着一定差别:16S采用的是将16S rDNA与Greengene(或silva)数据库进行比对注释,只能注释到细菌;而宏基因组则是将预测得到的基因与NR数据库比对从而进行注释,宏基因组注释得到的物种信息更为全面,不仅包括细菌,还包括真菌、古菌以及病毒等
- 此外,16S扩增子和宏基因组分析得到的注释结果也会存在一定的相似点,比如在门水平上相对丰度排名靠前的物种的类别会出现相似等情况;
综上所述,两者的分析方法本身存在一定的差异,是导致16S扩增子和宏基因组分析得到的注释结果存在差别的主要原因,但同时两者也有一定的相似之处。
个人经验实例:两者在细菌有多大差别?下面举一个我同学海哥的分析实例,对某样品同时进行16S和metagenome,其中展示了细菌中丰度大于1%的菌属种类,16S有15个属,metagenome有14个属,两者共有只有3个属,用黄色高亮显示。
16S by QIIME taxonomy greengene
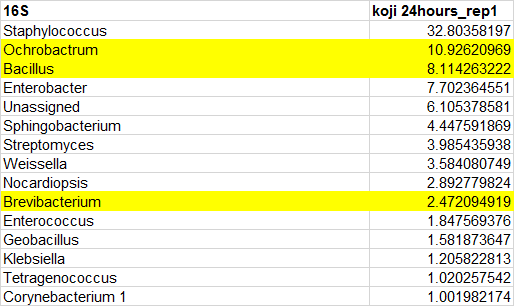
Metagenome by Metaplan2

个人感觉差异原因主要来自测序目标、技术方法、分析软件及数据库均不同。因为很多文章在Taxonomy水平更多使用16s的结果,而功能注释KEEG/COG则使用metagenome的结果。
03 宏基因组组装中,为什么不能把所有样本数据合并在一起进行组装?
不同样本中高丰度物种的差异很大,如果把所有样本都混合在一起进行组装,将会大大增加数据的复杂度,组装效果可能会更差。
04 在组装过程中,是否是共有的高丰度基因可以组装出来,而个体特有的低丰度的基因不能组装出来?
1)由于受到测序深度及测序成本的影响,在现在的宏基因组文章中,测序数据量一般选择6G,可以测出样品中绝大多数的微生物,但是对于一些低丰度的物种,因为测序深度的原因,确实很有可能会组装不出来;
2)在宏基因组分析中,也一般多关注的是较高丰度物种的组成情况,如果要对低丰度物种进行特殊分析,一般需要加大测序数据量,或者在前期提取过程中经过一些特殊的处理,尽可能的富集出多的低丰度物种,再进行测序分析。
个人经验:6G数据只适合简单系统,如人类肠道等,对于复杂系列,如土壤,致使测序几十到几百G,也可能也会深度不足。
05 宏基因组测序是否可以对抗性基因相关性进行分析,所用数据库是什么?
随着人们对抗性基因相关研究的广泛关注,我们宏基因组的标准分析中推出了抗性基因的相关分析。并且,由于自2009年ARDB数据库再无更新,因此我们目前所用的抗性基因数据库为CARD数据库。
声明
文章的解读仅代表个人理解和观点,有不足处,请读者积极留言批评指正,互相学习,共同进步。
图中大部分引用内容已经注明了原文链接,如有或遗漏或侵权请联系我 wechat: yongxinliu,谢谢。