【前言】
从本期起,小编将带领大家使用殷赋云计算平台(http://cloud.yinfotek.com/)来做一系列计算“文章”。这一期咱们来重现一篇2018年文献[1]的计算结果,看看作者是如何运用分子对接技术来阐明机理、升华文章的。该文献影响因子4.6,是典型的“实验+计算”模式,比较接近大多数科研人员的研究现状,只要熟悉这种模式,稍加努力即可发表同等水平的论文。
【研究背景】
文献作者通过细胞MTT试验从8000多个小分子中发现了一个新型的抗流感病毒化合物D715-2441。进一步实验发现,它对流感A病毒多种亚型(H1N1、H5N1、H7N9、H3N2、临床分离株690(H3)和带H274Y NA变异的奥司他韦抗性株)均有抗病毒活性,能够抑制病毒复制的早期阶段。更重要的是,D715-2441明显抑制病毒聚合酶的活性,并且直接影响PB2蛋白的定位。结合亲和力分析明确了该化合物特异性结合在PB2cap蛋白上。为了阐明抑制剂D715-2441的作用机理,文献作者采用DOCK 6分子对接技术分析其结合模式。
【计算步骤】
简单来说,分子对接的计算步骤包括4步:
1、 准备配体(小分子)和受体(生物大分子,比如蛋白)结构;
2、 定义对接口袋;
3、 进行对接计算;
4、 分析对接结果(打分与结合模式分析)。
下面,小编图文并茂地教大家使用殷赋云计算平台的DOCK6方案来完成对接计算与分析。本教程不打算详尽地讲解每一个操作步骤,仅将大体过程和关键要点讲清楚。但讲了很多值得注意的细节,这是一般教程上看不到的经验之谈。读者如果对本平台操作不是很熟悉,请翻看相应教程(分子对接结合模式预测教程)。
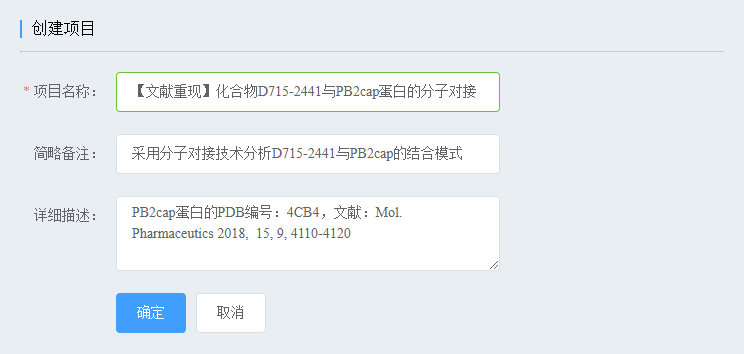
1、 创建项目和任务
登录平台,创建项目,填写内容。“简略备注”和“详细描述”是给你自己看的,可不填,但填写备忘是个值得鼓励的好习惯。然后,选择计算方案或工具。在这里,我们选择跟文献同款的Dock6(结合模式预测)方案。之后,填写任务内容。
2、 准备配体分子
平台为准备配体分子提供了多种输入方式,最简单的莫过于在线画结构(绘制小分子)。在画板上画出D715-2441的化学结构,并填上分子名称(不要有中文、空格、特殊符号或除英文字母外的其他语言文字!)和点击添加按钮。

输入的分子需要经过处理才能用于对接,其中包括:添加氢原子、添加电荷、生成3D结构和能量最小化。平台已对大多数情况进行优化设计,因此,采用默认设置即可。
值得注意的是:1、MMFF94力场是目前针对有机小分子最好的力场之一,也包括一些金属元素[2],但并非化学元素周期表全部元素都支持,必要时请使用UFF力场;2、电荷和能量优化使用的力场最好一致。
点击准备按钮。
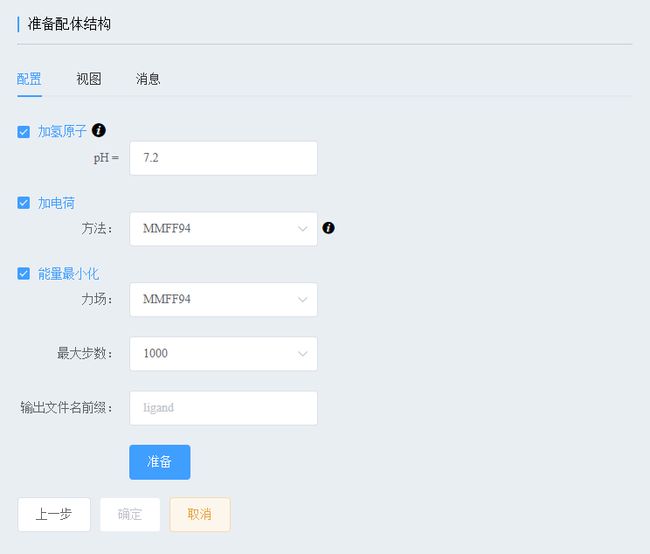
我们从视图中看到经过处理的配体结构。尽管平台已经做得很好,但人工检查仍是必要的。请根据你的化学知识,仔细检查结构是否正确,比如价键是否平衡、结构是否无扭曲、构型是否正确。确定无问题后,点击确定按钮;否则,返回前面,重新画结构或采用其他输入方式。
3、 准备受体分子
与配体不同,受体分子通常是生物大分子,结构大而复杂,无法手绘准备。通常采用实验解析的结构,可从RCSB 蛋白数据库下载。本工作用到的PB2cap蛋白PDB编号是4CB4。平台提供了两种输入方式:1)输入PDB ID;2)上传PDB文件。这里采用第一种方式。

通常来说,从PDB库下载的晶体结构文件里边包含了很多组分,不仅有(单条或多条)肽链,通常还有有机小分子和溶剂分子(水分子、金属离子)。如果下载的是NMR结构,还可能包含多个蛋白构象(称为model)。
在准备受体结构这一步,我们需要定义选用哪个模型(默认为1)、哪部分结构归为受体、哪部分为配体(将被提取出来用于口袋定位;若没有,可不定义)和其他杂质。在这里,我们选择A:PROTEIN为受体,A:MGT1484为配体,没有选上的部分将在后续处理中被程序删除。

4、 定义对接口袋
我们需要定义化合物D715-2441结合的大致区域——对接口袋。在上面的步骤中,我们提取了晶体结构中原先就存在的配体分子。通过文献了解到,它的结合位置就是D715-2441可能的结合区域。事实上,对该蛋白而言,也只有这里才靠谱。
通过选择文件方式,从弹出的文件列表选择MGT.pdb,点击显示盒子。稍等片刻,视图中将出现一个绿色盒子将MGT分子包围。我们需要保证的是,该盒子足够大,能够把口袋包围住。默认设置基本足够,必要时可增大小球范围(可能导致盒子扩大)。然后点击生成格点。这一步通常比较久,请耐心等待。然后,点击确定按钮。

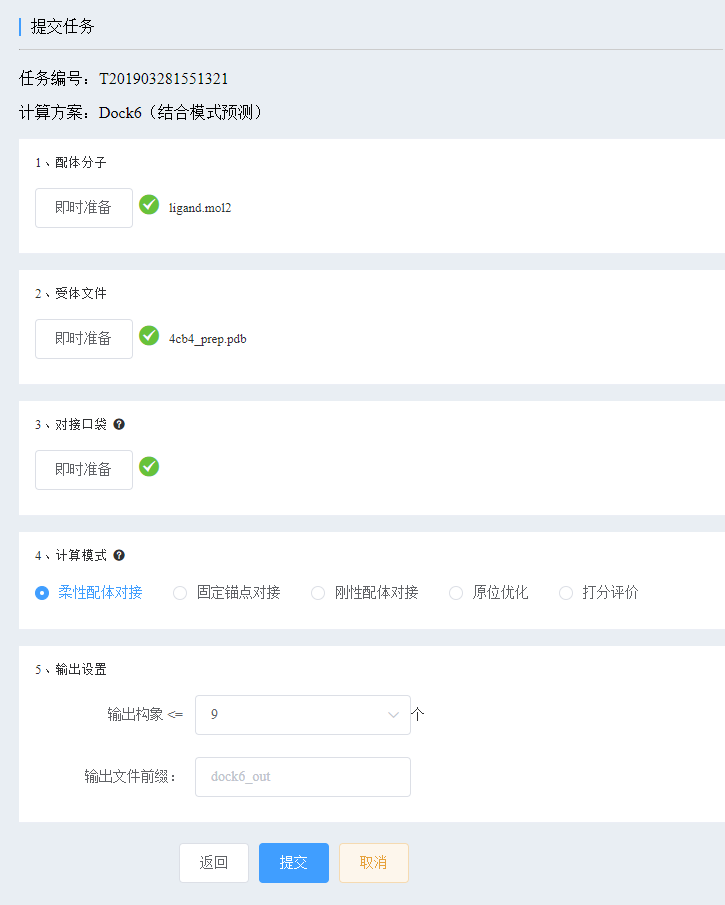
5、 提交计算任务
Dock6方案为我们提供了多种对接计算模式,这里稍微解释一下。
在各种模式中,蛋白受体都是刚性不动的。因为Dock6的Grid算法实际上并非直接采用蛋白结构进行对接,而是采用其模型(正是前面生成的格点)。根据配体运动的方式,分为下面几种情况:
1) 柔性配体对接,即配体在对接过程中是柔性可变的,可旋转单双键产生不同构象。这是最常用的默认的模式;
2) 固定锚点对接,即在口袋中对配体进行小范围构象搜索,配体有一块结构(通常为最大的刚性结构)看似被固定住,而其他部分则可变动。该模式的用处是,在保持配体整体结合位置不变的情况下,寻找更好的结合模式;
3) 刚性配体对接,即配体是刚性的,不能旋转化学键,仅可整体平移和翻转。可能的使用场景是,配体的构象具有某种特殊意义,希望对接后保持不变,或者评估蛋白是否能够容纳这种构象;
4) 原位优化(in-situ minimization),即在口袋中对配体分子进行能量优化,不进行构象搜索。可用于优化配体的结合模式;
5) 打分评价,即对口袋中的配体分子进行打分评价,不进行构象搜索。可采用不同打分函数对配体某一结合模式进行重打分。
我们采用柔性配体对接模式,其他参数采用默认值,然后提交任务。
6、 对接结果分析
1) 查看对接打分
Dock6方案的对接总打分是Grid Score,负值表示有结合,正值表示不结合,因此,打分值越小(负值的绝对值越大)表示结合力越强。它与另外两项能量项——范德华力和静电力——的关系是:Grid Score = Grid_vdw + Grid_es。Internal Energy是分子内能,表示对接构象(pose)的张力大小,正值,越小越好,通常20 kcal/mol以下比较合理。Pose列是对接构象编号,按照Grid Score从小到大排列。因此,第一个pose即为对接软件根据打分判断为最好的对接构象。
根据经验,Grid Score>-40 kcal/mol属于结合力较差,-40~-50kcal/mol属于中等,<-50kcal/mol属于较好。由于D715-2441分子较小,所以疏水作用面积小,范德华力贡献自然较小,静电力贡献通常也不会很大,导致整体打分较差。这是可以理解的,也应理解为该对接构象还算合理。
从打分分布看,各个pose的打分相差不大,范德华力贡献为主,第一个pose的静电力贡献相对其他pose出奇地大,意味着它具有较多较强的静电作用力(比如氢键和盐桥)。

2) 分析相互作用
仔细观察各个pose,结合上面的对接打分。我们认为第一个pose 的作用力最多,结合力最好,结合方式符合预期,是最好的构象。对比文献,这个pose与之几乎完全一致(下面会解释差异之处)。因此,我们选择它来做结合模式分析。
为清晰起见,点击视图右侧Protein下的cartoon按钮,去掉cartoon样式。点击Ligand下的center按钮,居中显示配体。我们清晰地看到,化合物D715-2441的芳环结构夹在H357的咪唑环和F404的苯环之间形成三明治结构,并与之分别形成π-π堆积作用(黄线),还有与F404的疏水作用(灰线),这些作用力一起构成了范德华力,提供了约-35.48kcal/mol的结合力贡献。另一方面,E361的羧基和K376的质子化N原子同时与苯环上的羟基形成氢键,氢键距离、角度可从下方表格查到。F404骨架上的氧原子也与D715-2441另一个羟基形成氢键作用。与文献不同的是,K339的N+原子和H357的咪唑基都跟环酯基形成盐桥作用。这种作用通常认为是非典型的,所以文献未提及,但在我们的检测分析工具里,它被呈现出来。这些极性作用为化合物的结合提供了较强的静电力贡献(约-7.69kal/mol),这也是该pose相对其他pose而言静电力非常显著的原因。

跟文献存在差异的另一个地方是,文献显示Phe323与化合物存在π-π堆积作用,Phe325存在疏水作用。但在我们的分析结果中,并没有出现这两个作用力。实际上,这两个氨基酸残基距离底物稍远,刚好超过该分析工具的相互作用力几何判断标准。但若将其原子结构显示出来,即发现也在附近(下图)。在柔性配体对接模式下,蛋白是刚性的,蛋白口袋的形状、残基位置未必是与配体最契合的。普遍认为,蛋白-配体之间存在“诱导契合”效应。在真实情况下,由于分子热运动,蛋白、配体间距离不可能一成不变。对接计算只是展示了其静态的一面,在真实世界里,几何距离在临界点附近的作用力可能“若隐若现”。

【下期预告】
高质量的文章需要高质量的Figure。小编将在下期教大家使用PyMOL做上乘的蛋白-配体结合模式图。敬请关注~
【参考文献】
[1] Teng et al. A Small-Molecule Compound Has Anti-influenza A Virus Activity by Acting as a “PB2 Inhibitor”. Mol. Pharmaceutics, 2018, 15 (9), pp 4110-4120. DOI: 10.1021/acs.molpharmaceut.8b00531
[2]https://open-babel.readthedocs.io/en/latest/Forcefields/mmff94.html#mmff94-force-field
更多资讯,请登录www.yinfotek.com或关注微信公众号“殷赋科技”。