这个推送主要是系统发育树相关内容(内容很多,但这篇文章并不包含多基因级联建树,挖个坑下次写。我将一些tips和操作示例写在了可以划动的模块里(只能在公众号里划动,粘贴不来),让整篇推送看起来结构清晰,如果不想看可以跳过划动部分)。上一个推送的多序列比对(【陪你学·生信】十、编辑对多序列比对结果【陪你学·生信】九、多序列比对-Multiple Sequence Alignment(MSA))是系统发育分析的基础。
文章有点长,先放个流程图便于理清思路。

一、系统发育树能分析什么
系统发育分析是根据生物核苷酸/蛋白序列的相似程度(进行MSA)将其归结成分支和簇,从而推测一组基因或蛋白质或生物间的进化关系。当然,系统发育分析的前提是进化论。
实验中系统发育分析常见目的有:物种鉴定,基因功能鉴定,追溯基因起源等。
二、准备序列数据时需要考虑的问题
1. 建什么树?用什么序列?
(1)核苷酸序列nt还是氨基酸序列aa
系统发育分析所选的序列应包含足够的信息,全基因组序列包含信息多,但是全基因组序列的分析麻烦、耗时长;且有的时候研究对象就是某一个基因或者蛋白。所以多数情况下,我们使用基因或蛋白序列建树。
可以的话,选择的这个基因在基因组的拷贝数最好为1,这样避免了旁系同源基因建树的影响。有的生物基因组小,比如病毒,建立物种树有时可以用全基因组序列(nt)进行系统发育分析(如果是蛋白序列同源性分析当然是aa序列,如果是非编码区序列分析当然是nt序列)。
对于可以用aa或者nt序列分析的实验材料,没有标准的答案。如果DNA序列之间的一致度高于70%或进化距离很近用nt序列更合适,因为nt序列的比对已经很整齐了,还可以保留同义突变;如果亲缘关系较远,用aa更合适,MSA时可以使保守区域对齐。也可以两个都试一试,可以加入已知物种/进化关系的同源分子做对照,看哪个结果更符合已知事实。
之前看过一篇论文,说目前发表的文章中构建的系统发育树多数都重复不出来。建树很“主观”,受到序列的选择、MSA、比对后剪辑、建树算法和参数等影响。所以在实验得到一个相对准确和满意的发育树后,最好记录下操作过程和参数。
(2)做的是基因树 (gene tree) 还是物种树 (species tree)
同源基因是指由共同祖先基因衍生的基因,主要有三种关系:直系同源、旁系同源、异源同源。
如果在建树时选择了一个生物中的旁系同源基因,或者选择的是不同物种中的同源基因,即直系同源,那么构建的都属于基因树。
我们视生殖隔离的产生为物种分化的标志,所以基因分化时间和物种分化时间不一致。基因树和物种树主要存在的差异有:①两物种的两个基因分化发生在物种形成事件之前,导致对系统发育树中最长分支的分析过估计;②基因树的拓扑结构可能和物种树存在差异。
基于上述原因,只能通过基因树来推测物种树。研究人员构建物种树时往往利用多个基因或蛋白级联建树。

(3)残基替换饱和度检验(Test of substitution saturation)
生物在进化发育中,nt/aa序列的残基会发生替换,通过对nt/aa残基替代数计算获得进化距离,而系统发育树是在进化距离基础上构建的。所以有必要对即将构建进化树的序列进行替换饱和度检验。
替换饱和度指的是一条序列同一位点残基发生多次替换(替换了几次又变成一样的残基了),或不同序列发生相同的替换。这会导致原本应有很大差异的两个序列,计算后却进化距离很近,从而导致长枝吸引现象(long branch attraction ,LBA)。
如果序列替换趋向于饱和,那么包含的系统发育信息少,用这些序列建树的意义不大。很多时候我们直接建的树感觉没啥大问题,常忽略饱和度检验。

(4)其他建树序列选择的tips
避免使用不完整的序列片段开始多序列比对;
避免使用异源同源序列;
避免使用重组序列。

2. 多序列比对结果编辑
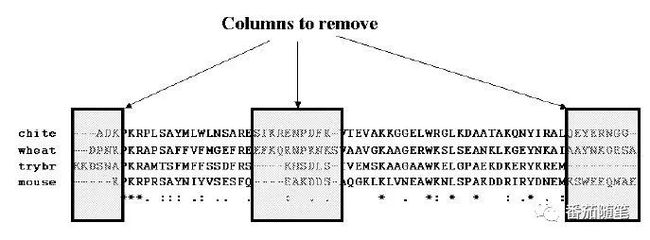
多序列比对结果的质量是影响建树质量的关键因素。应使其尽量:
(1)没有大量gap
(2)剪辑MSA不齐的两端
(3)保守区域选择:即优化MSA质量,保证比对结果中含有信息量大的板块,使比对质量高,但是残基一致性低。适用于信息位点足够多的长序列。
根据以上原则(1)(2),图片中短序列MSA结果中三个框的部分就可以删除。

三、构建系统发育树
1. 不同建树方法
建树原理主要分为基于距离的方法,即输入距离矩阵即可的距离法;以及基于序列信息的方法,即简约法、似然法等。常见的方法简单介绍如下(更详细的原理有空再写,挖个坑):
(1)邻接法(Neighbor-Joining,NJ):基于最小进化原理,通过比较每对序列的距离,构建一个总距离最小的拓扑树。速度最快,结果较为准确,比较常用;更适用于进化距离不大(0<平均距离<1),信息位点少的短序列。
(2)最大简约法(Maximum parsimony,MP):基于进化过程中残基替代数目应为最少的假说,即寻找一个最简约的含有最少转换事件/核苷酸替换/氨基酸替换的拓扑结构。不需要替换模型,更适用于序列残基差别小、具有近似变异率、信息位点较多的长序列。
(3)最大似然法(Maximum Likelihood,ML):对所有可能系统发育树的似然函数进行计算,即选定一个替代模型分析一组序列数据,选择似然率最大的树。在进化模型选择合适的情况下,ML结果可与进化事实很好吻合。目前最常用,计算强度大时速度慢。
(4)贝叶斯法(Bayesian Inference,BI):基于进化模型的统计推论法。可以处理大而复杂的数据集,将现有系统发育结论作为先验概率,通过后验概率直观反映各分支的可靠性而不需要通过自举法检验。

2. 系统发育树的评估
常用进化树的检验用的是自展检验(Bootstrap method)法,检验次数一般1000次,可将分支上的百分数视为这支结果的可信度。如果phylogeny test选择是None的话,分支上就没有数字。一般低于50是不太可信的,有人也说需要大于70,根据自己的实验具体分析。
美化树时也可以对步长值小于某个固定值的数字进行隐藏或者根据步长值对树进行修改。

3. 看懂系统发育树
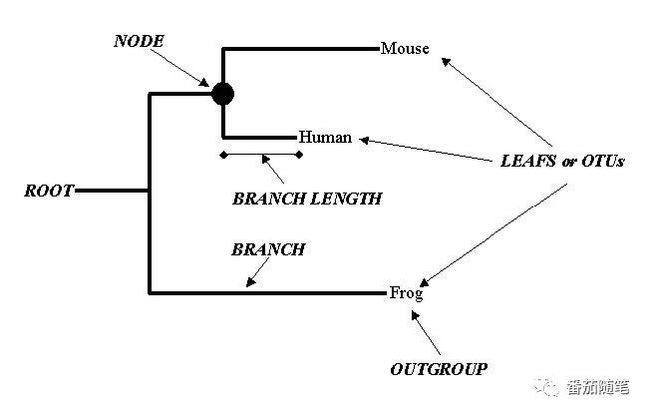
从图片右到左:
进化枝/操作分类单元(leafs/OTUs(Operational Taxonomic Units)):表示输入的序列;
进化分支(branch)表示这个枝上的OTU和其他序列的进化关系;
分支长度(brance length)在有标尺的树中可表示进化距离;
结点/节点(node)可表示非输入的/可能的祖先序列;
根(root)是共同祖先序列位置,可以引入外群构建有根树,即与你待分析序列关系密切且能很好的聚为一支(若外群不止一条序列)的序列。若研究演化,一般选择比目标序列具有较早进化历史的序列作为外群。root应出现在外群序列和其他分析序列的分支相连位置;
无根树(unrooted tree),无根树中的任何node都可能是距离原始序列最近的点,只表示这一组序列之间的相对进化关系。或者有时一组没有外群的序列建树也产生了“根”,但它可能只是建树上的“祖先”,并非具有准确生物学意义的祖先,因为系统发育树并不能确切指示进化方向,分析时不要混淆。
标尺(scale),不同算法,标尺的意义不同。NJ中表示遗传距离;MP中表示性状状态变换的步骤数,ML和BI中表示每个位点上的替换数。
四、系统发育树的美化
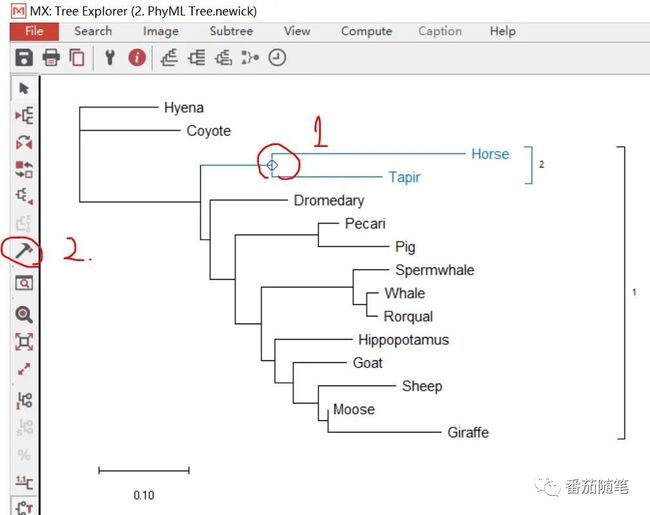
首推MEGA-X建树后的美化功能,可以给系统发育树不同的簇标色(也可以复制到word/PPT中简单编辑)。
先选中一个node,然后点击左侧的小锤子,可以给这个结点之下的分支标色,右侧也会有括号2,这个具体使用时,“2”可以是类/簇/不同寄主/不同界门纲目等的名称。
还有一个线上美化网页iTOL:
https://itol.embl.de/
以及需要下载的FigTree小软件:
http://tree.bio.ed.ac.uk/software/figtree/

五、一些在线资源
除了MEGA-X等本地软件,还有一些线上的资源,比如CLUSTAL多序列比对网站可以建树,还有Phylip,PhyML等。
如果你的数据复杂,用本地软件,可能很久都不能关机。这时候这些线上工具的优势就来了!提交数据,留下邮箱,就可以关机啦。
不过有的服务器对序列文件有限制,大数据无法在线操作分析,这时只能选择本地。
还有个很全的工具箱网站推荐:
https://molbiol-tools.ca/Phylogeny.htm

你们看sci-hub在飘雪花
很开心在这段拖更的日子里居然涨粉了!
拖更是因为,毕竟实验嘛,又到了年底冲业绩的时间啦,嘿嘿。

往期相关内容:
【陪你学·生信】序
【陪你学·生信】一、生信能帮我们做什么
【陪你学·生信】二、一些你肯定会用到的生信工具和基本操作
【陪你学·生信】三、核苷酸序列数据库的使用
【陪你学·生信】四、蛋白质相关的数据库
【陪你学·生信】五、当你有一段待分析的DNA序列(基础操作介绍)
【陪你学·生信】六、当你有一段待分析的氨基酸序列(基础操作介绍)
【陪你学·生信】七、在数据库中检索相似的序列
【陪你学·生信】八、序列两两比对
【陪你学·生信】九、多序列比对-Multiple Sequence Alignment(MSA)
【陪你学·生信】十、编辑对多序列比对结果