1.jellyfish
安装jellyfish
$ wget http://www.cbcb.umd.edu/software/jellyfish/jellyfish-1.1.10.tar.gz
$ tar zxvf jellyfish-1.1.10.tar.gz
$ mkdir jellyfish
$ cd jellyfish-1.1.10
$ ./configure --prefix=$PWD/../jellyfish
如果安装在当前目录中,会报错。
$ make -j 8
$ make install
运行jellyfish
cp denovo/newBGIseq500* .
gunzip newBGIseq500_1.fq.gz
gunzip newBGIseq500_2.fq.gz
#第一种方法
program/jellyfish count -m 17 -o kmer17 -s 268435456 -t 3 -c 8 -C newBGIseq500_1.fq newBGIseq500_2.fq
program/jellyfish stats -o kmer17.stats kmer17_0
program/jellyfish histo -t 3 kmer17_0 > kmer17.histo
less kmer17.histo | awk '{sum +=$2*$1}END{print sum/41}'
#6.55551e+06
cat kmer17.stats kmer17.histo |perl -lane '{if(/Distinct:\s+(\d+)/){$total=$1/100}elsif(/\d+\s+(\d+)/){print "$1\t".($1/$total);}}' >kmer_freq_distribution
#画图
Rscript kmer_plot.R
`
png("kmer_distibution.png")
a <- read.delim("kmer_freq_distribution",head=F)
l<-seq(1,nrow(a),by=1 )
plot(x=l,y=a[,2],type ="l",col ="red",lwd=2,xlim=c(0,200),ylim=c(0,1.5),main="kmer频率分布图",xlab="depth",ylab="Frequency(%)")
#text(40,0.05,"38")
dev.off()
`
#第二种方法
program/jellyfish count -m 17 -o 17k1 -s 268435456 -t 3 -c 8 -C newBGIseq500_1.fq
program/jellyfish count -m 17 -o 17k2 -s 268435456 -t 3 -c 8 -C newBGIseq500_2.fq
program/jellyfish merge -o 17kk.jf 17k*
diff 17kk.jf kmer17_0
#2个文件一样,没有区别
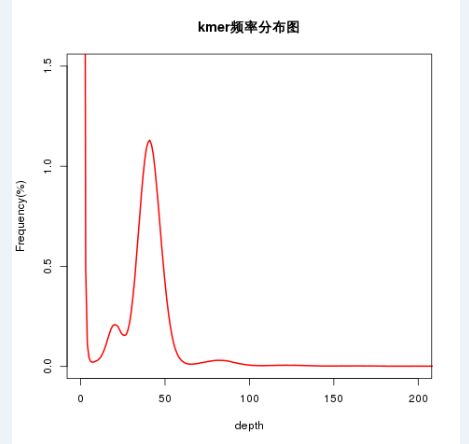
image.png
2.使用 GCE 进行基因组大小评估
gce-1.0.0/kmerfreq/kmer_freq_hash/kmer_freq_hash \
-k 17 -l read_list -i 450000000 -p species &> kmer_freq.log
gce-1.0.0/gce -f species.freq.stat -c 41 -H 1 -g 268799832 -m 1 -D 8 > species.table 2> species.log
下载GCE
wget ftp://ftp.genomics.org.cn/pub/gce/gce-1.0.0.tar.gz
tar zxf gce-1.0.0.tar.gz -C /opt/biosoft
GCE 软件包中主要包含 kmer_freq_hash 和 gce 两支程序。前者用于进行 kmer 的频数统计,后者在前者的结果上进行基因组大小的准确估算。
kmer_freq_hash 的常用参数:
Usage: kmer_freq_hash [options]
-k k-mer size(9~27), default=17
-l set read file list
-c set min accuracy rate of k-mer, set between 0~0.99, or -1 for unrestrained, default=-1
-q set Quality value ascii shift, generally 64 or 33, default 64
-r read length used to get k-mers, default=read's real length
-a ignored length of the beginning of a read, default=0
-d ignored length of the end of a read, default=0
-g total bases used to get k-mers, default=all input bases
-i initial size of hash table, default=1048576
-p set output prefix, default=output
-o set whether ouput k-mer frequence file, 0 for no, 1 for yes, default=1
-t thread number, default=1
-L maximum read length, default=100
-h output help information to screen
Example:
kmer_freq_hash -k 17 -i 450000000 -l fq.lst 2>kmerfreq.log;
kmer_freq_hash -k 19 -L 150 -i 600000000 -c 0.9 -t 8 -l fq.list 2>kmerfreq.log;
Attension: Please don't set -d and -r at the same time.
-k <17>
设置 kmer 的大小。该值为 9~27,默认值为 17 。
-l string
list文本文件,其中每行为一个fastq文件的路径。
-t int
使用的线程数,默认为 1 。
-i int
初始的 hash 表大小,默认为 1048576。该值最好设置为 (kmer 的种类数 / 0.75)/ 线程数。如果基因组大小为 100M,测序了 40M 个 reads,reads 的长度为 100bp,测序错误率为 1%,kmer的大小为 21,则kmer的种类数为100M+40M*100*1%*21=940M,若使用24线程,则该参数设置为 i=940M/0.75/24=52222222。
-p string
设置输出文件的前缀。
-o int
是否输出 k-mer 序列。1: yes, 0: no,默认为 1 。推荐选 0 以节约运行时间。
-q int
设置fastq文件的phred格式,默认为 64。该值可以为 33 或 63。
-c double
设置k-mer最小的精度,该值位于 0~0.99,或为 -1。 -1 表示不对 kmer进行过滤。设置较高的精度,可以用于过滤低质量 kmer。精度是由 phred 格式的碱基质量计算得来的。
-r int
设置获取 k-mer 使用到的 reads 长度。默认使用 reads 的全长。
-a int
忽略read前面该长度的碱基。
-d int
忽略read后面该长度的碱基。
-g int
设置使用该数目的碱基来获取 k-mers,默认是使用所有的碱基来获取 k-mer。
运行kmer_freq_hash:
gce-1.0.0/kmerfreq/kmer_freq_hash/kmer_freq_hash \
-k 17 -l read_list -i 450000000 -p species &> kmer_freq.log
kmer_freq_hash 的主要结果文件为 species.freq.stat。该文件有 2 列:第1列是kmer重复的次数,第二列是kmer的种类数。该文件有255行,第225行表示kmer重复次数>=255的kmer的总的种类数。该文件作为 gce 的输入文件。
kmer_freq_hash 的输出到屏幕上的信息结果保存到文件 kmer_freq.log 文件中。该文件中有粗略估计基因组的大小。其中的 Kmer_individual_num 数据作为 gce 的输入参数。
less -S kmer_freq.log
**********Summary Table**********
Kmer_size Kmer_individual_num Kmer_species_num Filter_kmer_num Kmer_depth(coverage) Genome_size(rough) Base_num Base_depth(coverage)
17 268799832 25667758 0 41.2466 5993008 319999800 53.3955 3199998 100
#看不清
Kmer_size 17
Kmer_individual_num 268799832
Kmer_species_num 25667758
Filter_kmer_num 0
Kmer_depth(coverage) 41.2466
Genome_size(rough) 5993008
Base_num 319999800
Base_depth(coverage) 53.3955 3199998 100
$ head species.freq.stat
1 18626187
2 1256204
3 125673
4 28077
5 11014
6 6299
7 5330
8 5810
9 6887
10 7796
自己计算下Genome_size:peak=41
$ less -S species.freq.stat | awk '{sum += $1*$2} END {print sum/41}'
6.4344e+06 #这里是6.4M 比估算的6M要大
去掉第一行(1 18626187),计算为5980098,跟估算结果差不多
>a <-read.table("species.freq.stat")
>b <-a[,1]*a[,2]
> sum(b[2:length(b)])/41
[1] 5980098
gce 的使用:
gce-1.0.0/gce -f species.freq.stat \
-c 41 -g 268799832 -m 1 -D 8 > species.table 2> species.log
参数说明:
-f string
kmer depth frequency file
-c int
kmer depth frequency 的主峰对应的 depth。gce 会在该值附近找主峰。
-g int
总共的 kmer 数。一定要设定该值,否则 gce 会直接使用 -f 指定的文件计算 kmer 的总数。由于默认下该文件中最大的 depth 为 255,因此,软件自己计算的值比真实的值偏小。同时注意该值包含低覆盖度的 kmer。
-M int
支持最大的 depth 值,默认为 256 。
-m int
估算模型的选择,离散型(0),连续型(1)。默认为 0,对真实数据推荐选择 1 。
-D int
precision of expect value,默认为 1。如果选择了 -m 1,推荐设置该值为 8。
-H int
使用杂合模式(1),不使用杂合模式(0)。默认值为 0。只有明显存在杂合峰的时候,才选择该值为 1 。
-b int
数据是(1)否(0)有 bias。当 K > 19时,需要设置 -b 1 。
gce 的结果文件为 species.table 和 species.log 。species.log 文件中的主要内容:
raw_peak now_node low_kmer now_kmer cvg genome_size a[1] b[1]
41 5614870 21725832 242490544 40.8357 6.05044e+06 0.944654 0.844481
raw_peak: 覆盖度为 41 的 kmer 的种类数最多,为主峰。
now_node: kmer的种类数。
low_kmer: 低覆盖度的 kmer 数。
now_kmer: 去除低覆盖度的 kmer 数,此值 = (-g 参数指定的总 kmer 数) - low_kmer 。
cvg:估算出的平均覆盖度
genome_size:基因组大小,该值 = now_kmer / cvg 。
a[1]: 在基因组上仅出现 1 次的 kmer 之 种类数比例。
b[1]: 在基因组上仅出现 1 次的 kmer 之 数量比例。该值代表着基因组上拷贝数为 1 的序列比例。
如果使用 -H 1 参数,则会得额外得到如下信息:
for hybrid: a[1/2]=0.109694 a1=0.824146
kmer-species heterozygous ratio is about 0.0580297
for hybrid: b[1/2]=0.0680686 b1=0.77641
kmer-individual heterozygous ratio is about 0.0352335
Final estimation table:
raw_peak now_node low_kmer now_kmer cvg genome_size a[1/2] a[1] b[1/2] b[1]
40 5621531 21685380 242491591 40.8306 6.05219e+06 0.109694 0.824146 0.0680686 0.77641
上面结果中,0.0580297 是由 a[1/2] 计算出来的。 0.0580297= a[1/2] / ( 2- a[1/2] ) 。
a[1/2]=0.109694 表示在所有的 uniqe kmer 中,有 0.109694 比例的 kmer 属于杂合 kmer 。
此外,有 a[1/2] 和 b[1/2] 的值在最后的统计结果中。重复序列的含量 = 1 - b[1/2] - b[1] 。
则杂合率 = 0.0580297 / kmer_size 。 若计算出的杂合率低于 0.2%,个人认为测序数据应该是纯合的。这时候,应该不使用 -H 1 参数。使用 -H 1 参数会对基因组的大小和重复序列含量估算造成影响。
参考:https://www.plob.org/article/9388.html
3.KmerFreq_AR计算基因组大小

/home/zhaosl/biosoft/Assemblathon1_pipeline/KmerFreq_AR_v2.0 \
-k 17 -t 20 -p test read_list
ls test*
test.freq.cz test.freq.cz.len test.freq.stat test.genome_estimate
less -S test.genome_estimate
kmer kmer_num kmer_depth genome_size base_num read_num base_depth(X) average_read_len unique_kmer_number
17 268799832 41.2466 5993008 319999800 3199998 53.3955 100 25667758
kmer 17
kmer_num 268799832
kmer_depth 41.2466
genome_size 5993008
base_num 319999800
read_num 3199998
base_depth(X) 53.3955
average_read_len 100
unique_kmer_number 25667758