scATAC分析神器ArchR初探-简介(1)
scATAC分析神器ArchR初探-ArchR进行doublet处理(2)
scATAC分析神器ArchR初探-创建ArchRProject(3)
scATAC分析神器ArchR初探-使用ArchR降维(4)
scATAC分析神器ArchR初探--使用ArchR进行聚类(5)
scATAC分析神器ArchR初探-单细胞嵌入(6)
scATAC分析神器ArchR初探-使用ArchR计算基因活性值和标记基因(7)
scATAC分析神器ArchR初探-scRNA-seq确定细胞类型(8)
scATAC分析神器ArchR初探-ArchR中的伪批次重复处理(9)
scATAC分析神器ArchR初探-使用ArchR-peak-calling(10)
scATAC分析神器ArchR初探-使用ArchR识别标记峰(11)
scATAC分析神器ArchR初探-使用ArchR进行主题和功能丰富(12)
scATAC分析神器ArchR初探-利用ArchR丰富ChromVAR偏差(13)
scATAC分析神器ArchR初探-使用ArchR进行足迹(14)
scATAC分析神器ArchR初探-使用ArchR进行整合分析(15)
scATAC分析神器ArchR初探-使用ArchR进行轨迹分析(16)
16-使用ArchR进行轨迹分析
为了在伪时间内对单元进行排序,ArchR创建了细胞轨迹,该轨迹对ArchRProject中较低N维子空间上的单元进行排序。以前,我们已经在二维UMAP子空间中执行了此排序,但是ArchR在此方法上进行了改进,可以在N维子空间(即LSI)中进行对齐。首先,ArchR需要用户定义的轨迹主干,该主干可提供细胞组/簇的大致顺序。例如,给定用户确定的簇身份,可以提供干细胞簇的簇ID,然后是祖细胞簇,然后是与已知或推测的生物学相关细胞轨迹相对应的分化细胞簇(即提供簇) HSC,GMP,单核细胞的ID)。接下来,对于每个群集,ArchR计算N维中每个单元组/集群的平均坐标,并保留与这些平均坐标的欧几里得距离在所有单元的前5%中的单元。接下来,ArchR为每个单元计算从群集i到群集i + 1沿着轨迹的平均坐标的距离,并基于这些距离为i的每次迭代计算一个伪时间向量。这使得ArchR可以基于到单元格组/簇平均坐标的欧几里得距离,为作为轨迹一部分保留的每个单元格确定N维坐标和伪时间值。接下来,ArchR使用伪时间基于伪时间值将连续轨迹拟合到每个N维坐标。ArchR计算沿着轨迹从簇i到簇i + 1的平均坐标的每个单元的距离,并基于这些距离为i的每次迭代计算一个伪时间向量。这使得ArchR可以基于到单元格组/簇平均坐标的欧几里得距离,为作为轨迹一部分保留的每个单元格确定N维坐标和伪时间值。接下来,ArchR使用伪时间基于伪时间值将连续轨迹拟合到每个N维坐标。ArchR计算沿着轨迹从簇i到簇i + 1的平均坐标的每个单元的距离,并基于这些距离为i的每次迭代计算一个伪时间向量。这使得ArchR可以基于到单元格组/簇平均坐标的欧几里得距离,为作为轨迹一部分保留的每个单元格确定N维坐标和伪时间值。接下来,ArchR使用伪时间基于伪时间值将连续轨迹拟合到每个N维坐标。这使得ArchR可以基于到单元格组/簇平均坐标的欧几里得距离,为作为轨迹一部分保留的每个单元格确定N维坐标和伪时间值。接下来,ArchR使用伪时间基于伪时间值将连续轨迹拟合到每个N维坐标。这使得ArchR可以基于到单元格组/簇平均坐标的欧几里得距离,为作为轨迹一部分保留的每个单元格确定N维坐标和伪时间值。接下来,ArchR使用伪时间基于伪时间值将连续轨迹拟合到每个N维坐标。smooth.spline功能。然后,ArchR基于所有单元格到沿流形的最近点的欧几里德距离,将所有单元格与轨迹对齐。然后,ArchR将此对齐比例缩放为100,并将此伪时间存储在ArchRProject中以进行下游分析。
ArchR可以创建矩阵,以在Arrow文件中存储的要素之间传递伪时间趋势。例如,ArchR可以分析跨伪时间的TF偏差,基因得分或整合的基因表达的变化,以识别在整个细胞轨迹中动态的调节子或调节元件。首先,ArchR以用户定义的小幅度增量(默认= 1/100)在整个细胞轨迹上对细胞进行分组。然后,ArchR使用该功能使用用户定义的平滑窗口(默认值= 9/100)对每个功能对该矩阵进行平滑data.table::frollmean。然后,ArchR将此平滑的伪时间x特征矩阵返回为SummarizedExperiment用于下游分析。此外,ArchR可以使用名称匹配(即具有chromVAR TF偏差和基因得分/整合谱的正调控子)或通过基因组位置重叠法(即峰与基因的关联)使用低位相关性来关联这些平滑的伪时间x特征矩阵中的两个如前面部分所述,重叠的细胞聚集体。因此,ArchR促进了跨细胞轨迹的综合分析,揭示了跨多模态数据的相关监管动态。
16.1髓系轨迹-单核细胞分化
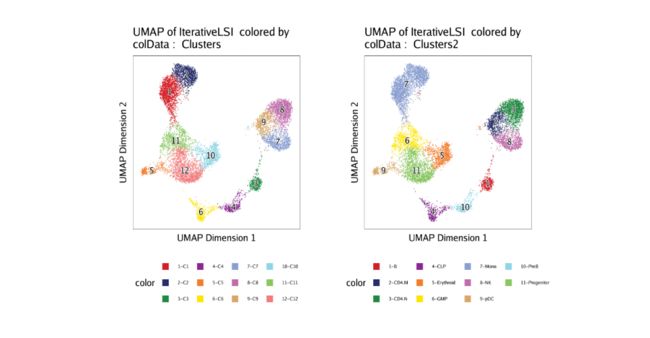
在本节中,我们将创建一个近似于HSCs向完全分化的单核细胞分化的细胞轨迹。首先,让我们检查一下我们先前定义的群集和单元格类型,它们存储在cellColData名为“ Clusters”和“ Clusters2”的列中。将这些单元格分组覆盖在我们的UMAP嵌入上会显示我们感兴趣的不同单元格类型。
p1 <- plotEmbedding(ArchRProj = projHeme5, colorBy = "cellColData", name = "Clusters", embedding = "UMAP")
p2 <- plotEmbedding(ArchRProj = projHeme5, colorBy = "cellColData", name = "Clusters2", embedding = "UMAP")
ggAlignPlots(p1, p2, type = "h")
16.1.1伪时间UMAP和单个特征图
我们将使用存储在“ Clusters2”中的单元格类型定义。如上所述,我们正在创建一条从干细胞(“祖细胞”)到定型的髓祖细胞(“ GMP”)到单核细胞(“单核”)的轨迹。创建轨迹的第一步是以细胞组标签的有序向量形式创建轨迹主干。
trajectory <- c("Progenitor", "GMP", "Mono")
trajectory
我们使用addTrajectory()函数创建轨迹,并将其添加到ArchRProject。我们将这个轨迹称为“ MyeloidU”。这样做是在cellColData“ MyeloidU”中创建一个新列,该列存储轨迹中每个单元的伪时间值。不属于轨迹的像元标记为NA。
projHeme5 <- addTrajectory(
ArchRProj = projHeme5,
name = "MyeloidU",
groupBy = "Clusters2",
trajectory = trajectory,
embedding = "UMAP",
force = TRUE
)
我们可以查看此信息,并看到每个像元都有一个介于0到100之间的唯一伪时间值。我们排除了具有NA这些值的像元,因为它们不是轨迹的一部分。
head(projHeme5$MyeloidU[!is.na(projHeme5$MyeloidU)])
要绘制此轨迹,我们使用在plotTrajectory()UMAP嵌入中覆盖伪时间值的函数,并显示一个近似于样条曲线拟合的轨迹路径的箭头。不属于轨迹的像元被涂成灰色。在此示例中,我们用于colorBy = "cellColData"告诉ArchR在cellColData由name- 指定的列(在本例中为“ MyeloidU”伪时间轨迹)内查找。虽然同时为trajectory和列出“ MyeloidU”似乎很反常name,但这是因为trajectory告诉ArchR我们感兴趣的是哪个细胞子集,并且name告诉ArchR如何为该细胞子集着色。
p <- plotTrajectory(projHeme5, trajectory = "MyeloidU", colorBy = "cellColData", name = "MyeloidU")
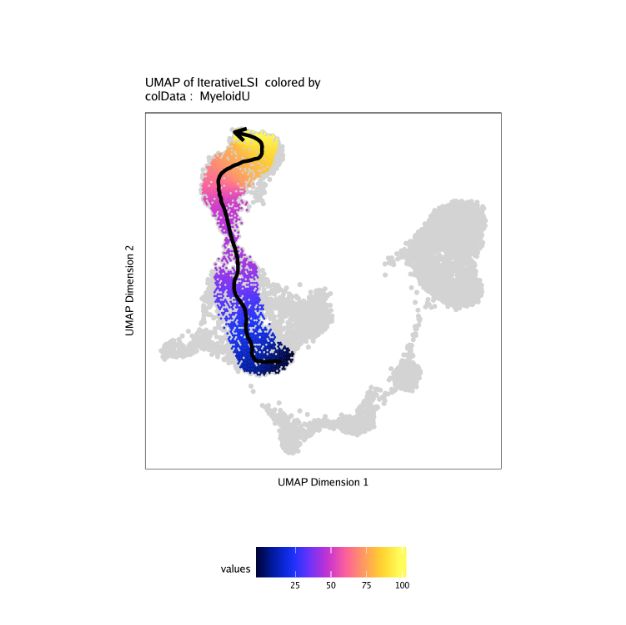
要保存此图的可编辑矢量化版本,请使用plotPDF()。
plotPDF(p, name = "Plot-MyeloidU-Traj-UMAP.pdf", ArchRProj = projHeme5, addDOC = FALSE, width = 5, height = 5)
我们还可以在UMAP嵌入中将其他要素叠加在轨迹上。这使我们仅在与我们的轨迹相关的像元内显示特定特征。
如果尚未将估算权重添加到projHeme5项目中,请立即执行。
projHeme5 <- addImputeWeights(projHeme5)
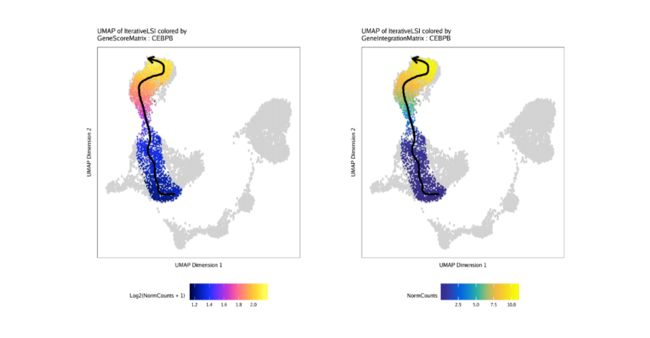
然后,我们可以绘制“ MyeloidU”轨迹,但通过CEBPB基因的基因得分值对细胞着色,CEBPB基因是已知的单核细胞功能调节剂,在分化过程中会活跃。我们指出要通过colorBy参数使用的矩阵和要通过参数使用的特征name。
p1 <- plotTrajectory(projHeme5, trajectory = "MyeloidU", colorBy = "GeneScoreMatrix", name = "CEBPB", continuousSet = "horizonExtra")
我们可以重复这个过程,但是通过细胞的连锁基因表达使细胞着色GeneIntegrationMatrix。
p2 <- plotTrajectory(projHeme5, trajectory = "MyeloidU", colorBy = "GeneIntegrationMatrix", name = "CEBPB", continuousSet = "blueYellow")
该plotTrajectory()函数实际上返回相关图的列表。列表中的第一个图是UMAP嵌入,按函数调用中的指定进行着色。
并排比较这些UMAP图的基因得分和基因表达,我们发现CEBPB基因的活性在伪时间轨迹的后期对单核细胞具有高度特异性。
ggAlignPlots(p1[[1]], p2[[1]], type = "h")
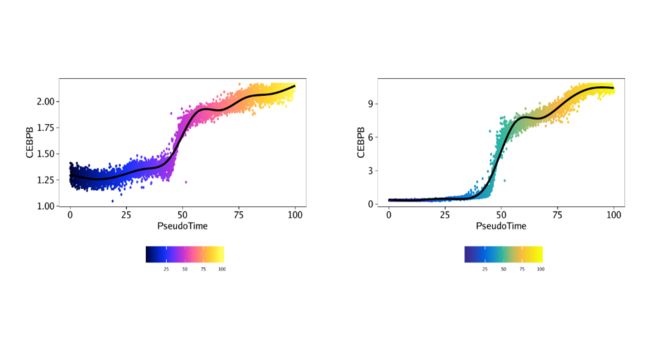
返回的第二个图是plotTrajectory()伪时间与相关特征值(在这种情况下为CEBPB的基因得分或基因表达)的点图。在这种情况下,单元格通过其伪时间着色。
ggAlignPlots(p1[[2]], p2[[2]], type = "h")
16.1.2伪时间热图
我们可以使用热图可视化跨伪时间的许多功能的变化。为此,我们首先ArchRProject使用getTrajectory()函数返回感兴趣的轨迹,该函数将轨迹作为SummarizedExperiment对象返回。通过将相应的矩阵传递给useMatrix参数,我们将为模体,基因得分,基因表达和峰值可及性创建这些伪时间热图。
trajMM <- getTrajectory(ArchRProj = projHeme5, name = "MyeloidU", useMatrix = "MotifMatrix", log2Norm = FALSE)
然后,我们将此传递SummarizedExperiment给plotTrajectoryHeatmap()函数。
p1 <- plotTrajectoryHeatmap(trajMM, pal = paletteContinuous(set = "solarExtra"))
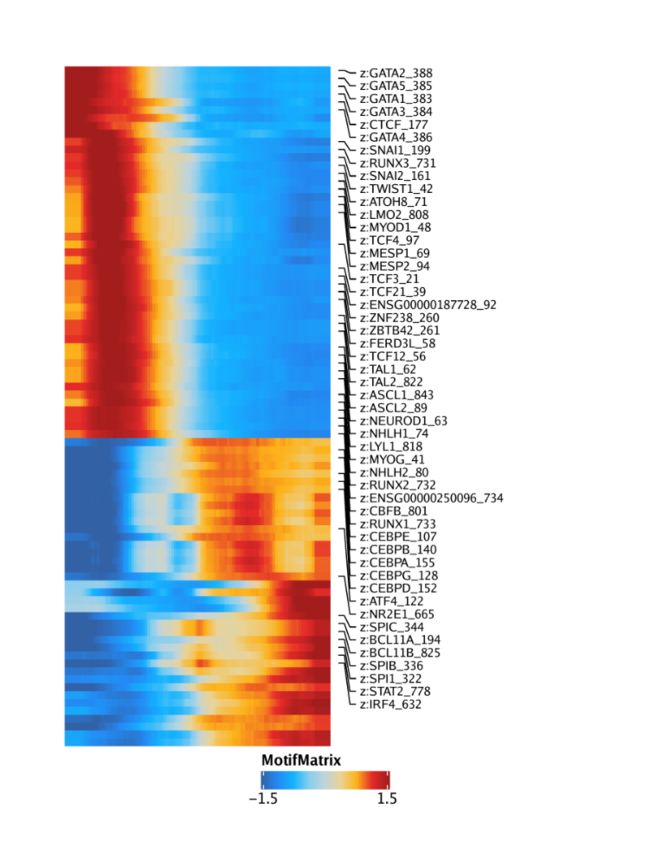
通过设置,我们可以执行相同的步骤来获得基因分数的伪时间热图useMatrix = "GeneScoreMatrix"。
trajGSM <- getTrajectory(ArchRProj = projHeme5, name = "MyeloidU", useMatrix = "GeneScoreMatrix", log2Norm = TRUE)
p2 <- trajectoryHeatmap(trajGSM, pal = paletteContinuous(set = "horizonExtra"))
同样,我们可以通过设置获得基因表达的伪时间热图useMatrix = "GeneIntegrationMatrix"。
trajGIM <- getTrajectory(ArchRProj = projHeme5, name = "MyeloidU", useMatrix = "GeneIntegrationMatrix", log2Norm = FALSE)
p3 <- plotTrajectoryHeatmap(trajGIM, pal = paletteContinuous(set = "blueYellow"))
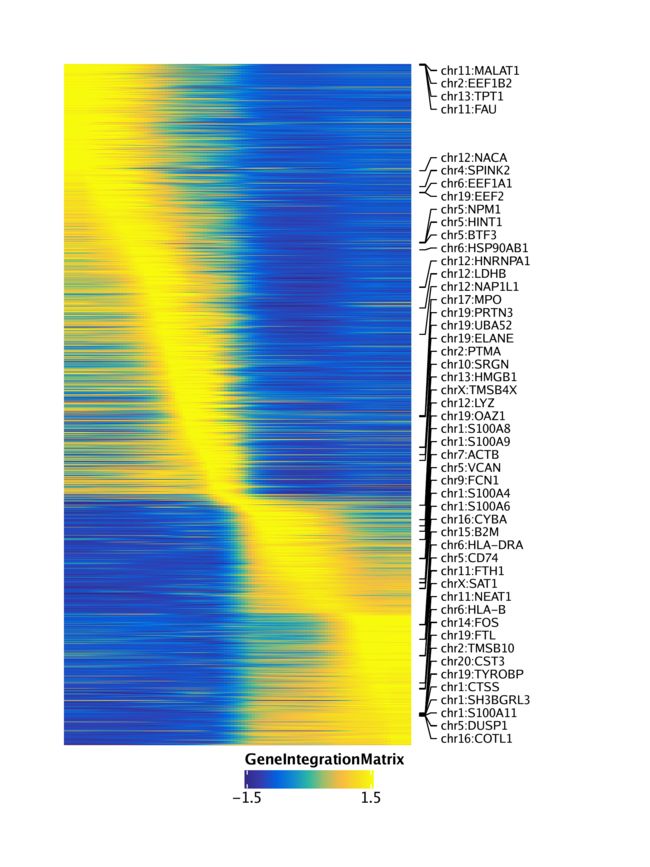
最后,我们可以通过设置获得峰值可访问性的伪时间热图useMatrix = "PeakMatrix"。
trajPM <- getTrajectory(ArchRProj = projHeme5, name = "MyeloidU", useMatrix = "PeakMatrix", log2Norm = TRUE)
p4 <- plotTrajectoryHeatmap(trajPM, pal = paletteContinuous(set = "solarExtra"))
要保存此图的可编辑矢量化版本,请使用plotPDF()。
plotPDF(p1, p2, p3, p4, name = "Plot-MyeloidU-Traj-Heatmaps.pdf", ArchRProj = projHeme5, addDOC = FALSE, width = 6, height = 8)
16.1.3集成伪时间分析
我们还可以进行综合分析,例如通过整合基因得分/基因表达和伪时间中的基元可及性来鉴定阳性TF调节剂。例如,在识别差异驱动因素时,这可能非常强大。为此,我们使用该correlateTrajectories()函数,该函数接受SummarizedExperiment从该getTrajectories()函数检索的两个对象。
首先,让我们找到其伪时间可访问性与TF基因的基因得分相关的基序。
corGSM_MM <- correlateTrajectories(trajGSM, trajMM)
的主要输出correlateTrajectories()是一个列表对象,其中包含一个DataFrame对象作为列表中的第一个条目。此DataFrame已命名的列idx1,matchname1,name1,和VarAssay1其对应于索引,匹配名,未改变的名字,并从第一轨迹的特征的方差位数传递给(基因分)correlateTrajectories()功能。“方差分位数”是对给定特征的归一化度量,它使我们能够得出不同分析之间的相关性。它DataFrame包含满足功能中指定的临界值的所有correlateTrajectories()功能。
corGSM_MM[[1]]
然后,我们可以将相应的轨迹SummarizedExperiment对象子集化,以仅包含通过上述意义的元素。
trajGSM2 <- trajGSM[corGSM_MM[[1]]$name1, ]
trajMM2 <- trajMM[corGSM_MM[[1]]$name2, ]
为了最好地排序这些功能,我们可以创建一条新的轨迹,将这两个轨迹的值相乘。这将使我们能够创建并排排列的热图,它们按行顺序相同。
trajCombined <- trajGSM2
assay(trajCombined) <- t(apply(assay(trajGSM2), 1, scale)) + t(apply(assay(trajMM2), 1, scale))
我们可以从plotTrajectoryHeatmap()函数的返回值中提取最佳行顺序。
combinedMat <- plotTrajectoryHeatmap(trajCombined, returnMat = TRUE, varCutOff = 0)
rowOrder <- match(rownames(combinedMat), rownames(trajGSM2))
这样,我们现在准备创建配对的热图。首先,我们将为基因得分轨迹创建热图。我们通过rowOrder参数指定所需的行顺序。
ht1 <- plotTrajectoryHeatmap(trajGSM2, pal = paletteContinuous(set = "horizonExtra"), varCutOff = 0, rowOrder = rowOrder)
然后,我们将为主题轨迹创建热图,再次通过rowOrder参数指定行顺序
ht2 <- plotTrajectoryHeatmap(trajMM2, pal = paletteContinuous(set = "solarExtra"), varCutOff = 0, rowOrder = rowOrder)
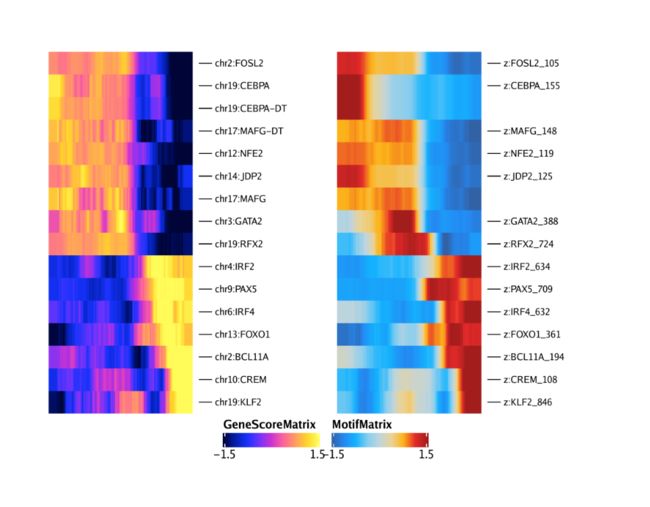
并排绘制这两个热图,我们看到在两个热图之间行是匹配的。您可能会注意到,该分析同时捕获了GATA3和GATA3-AS1(GATA3的反义转录本)。这是由于如何执行功能匹配,并且反义转录本条目可以在后期处理中手动删除,也可以根据需要通过编程方式删除。
ht1 + ht2
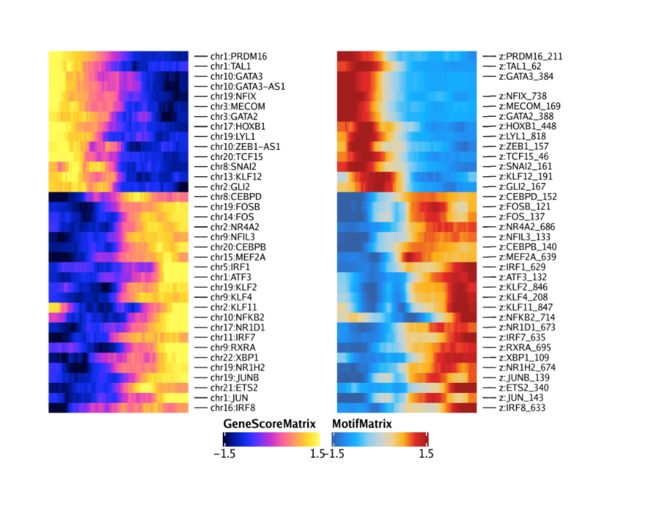
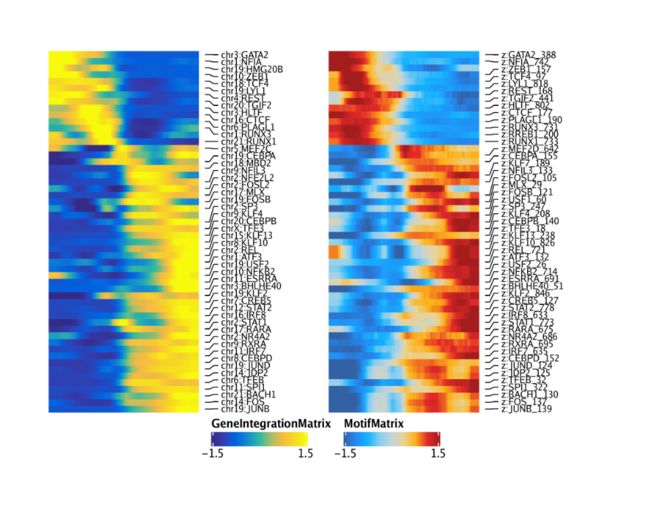
我们可以重复相同的确切过程,但是使用来自
GeneIntegrationMatrix而不是基因得分的基因表达。因为这是相同的分析工作流程,所以我们不会在每个步骤上重复说明。
corGIM_MM <- correlateTrajectories(trajGIM, trajMM)
corGIM_MM[[1]]
trajGIM2 <- trajGIM[corGIM_MM[[1]]$name1, ]
trajMM2 <- trajMM[corGIM_MM[[1]]$name2, ]
trajCombined <- trajGIM2
assay(trajCombined) <- t(apply(assay(trajGIM2), 1, scale)) + t(apply(assay(trajMM2), 1, scale))
combinedMat <- plotTrajectoryHeatmap(trajCombined, returnMat = TRUE, varCutOff = 0)
rowOrder <- match(rownames(combinedMat), rownames(trajGIM2))
ht1 <- plotTrajectoryHeatmap(trajGIM2, pal = paletteContinuous(set = "blueYellow"), varCutOff = 0, rowOrder = rowOrder)
ht2 <- plotTrajectoryHeatmap(trajMM2, pal = paletteContinuous(set = "solarExtra"), varCutOff = 0, rowOrder = rowOrder)
ht1 + ht2
16.2淋巴弹道-B细胞凋亡
作为轨迹的第二个例子,我们将从祖细胞到常见的淋巴样祖细胞和前B细胞一直到完全分化的B细胞,来创建B细胞轨迹。因为从上一节中的单核细胞轨迹开始基本上重复了此分析,所以我们不提供代码片段的解释。如果您想学习如何执行轨迹分析,请查看本章中的单核细胞轨迹部分。
p1 <- plotEmbedding(ArchRProj = projHeme5, colorBy = "cellColData", name = "Clusters", embedding = "UMAP")
p2 <- plotEmbedding(ArchRProj = projHeme5, colorBy = "cellColData", name = "Clusters2", embedding = "UMAP")
ggAlignPlots(p1, p2, type = "h")
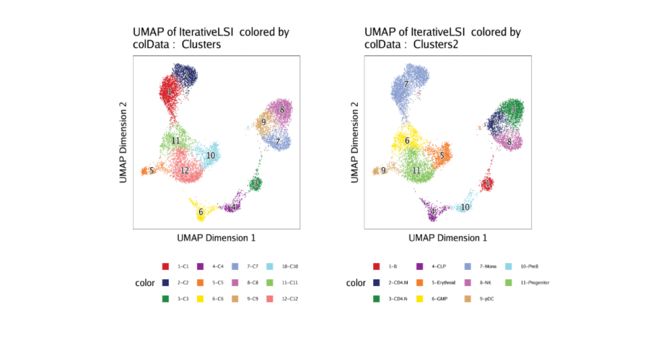
16.2.1伪时UMAP和单个特征图
trajectory <- c("Progenitor", "CLP", "PreB", "B")
trajectory
projHeme5 <- addTrajectory(
ArchRProj = projHeme5,
name = "LymphoidU",
groupBy = "Clusters2",
trajectory = trajectory,
embedding = "UMAP",
force = TRUE
)
head(projHeme5LymphoidU)]) #NA means not in trajectory
p <- plotTrajectory(projHeme5, trajectory = "LymphoidU", colorBy = "cellColData", name = "LymphoidU")
p[[1]]
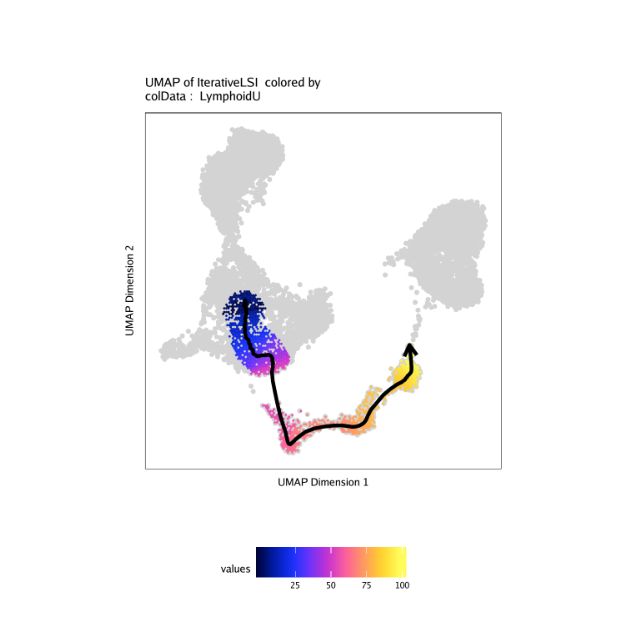
要保存此图的可编辑矢量化版本,请使用plotPDF()。
plotPDF(p, name = "Plot-LymphoidU-Traj-UMAP.pdf", ArchRProj = projHeme5, addDOC = FALSE, width = 5, height = 5)
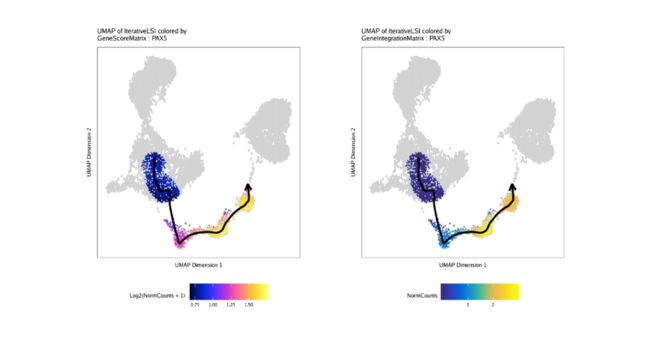
p1 <- plotTrajectory(projHeme5, trajectory = "LymphoidU", colorBy = "GeneScoreMatrix", name = "PAX5", continuousSet = "horizonExtra")
p2 <- plotTrajectory(projHeme5, trajectory = "LymphoidU", colorBy = "GeneIntegrationMatrix", name = "PAX5", continuousSet = "blueYellow")
ggAlignPlots(p1[[1]], p2[[1]], type = "h")
ggAlignPlots(p1[[2]], p2[[2]], type = "h")
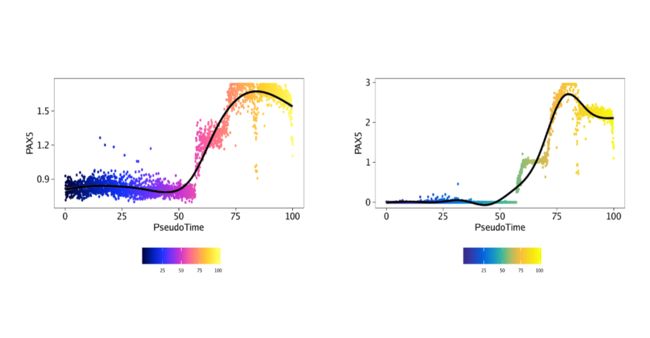
16.2.2伪时间热图
trajMM <- getTrajectory(ArchRProj = projHeme5, name = "LymphoidU", useMatrix = "MotifMatrix", log2Norm = FALSE)
p1 <- plotTrajectoryHeatmap(trajMM, pal = paletteContinuous(set = "solarExtra"))
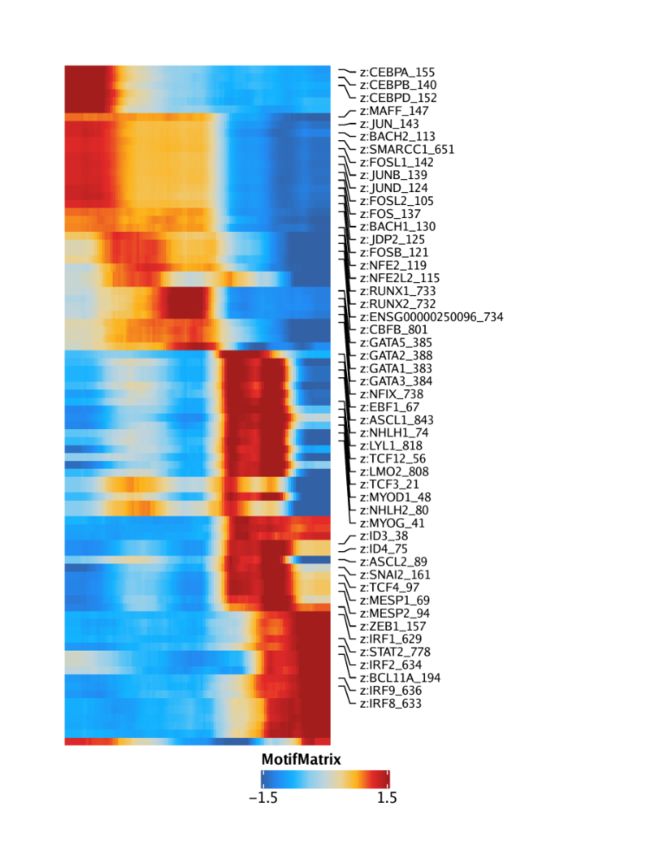
trajGSM <- getTrajectory(ArchRProj = projHeme5, name = "LymphoidU", useMatrix = "GeneScoreMatrix", log2Norm = TRUE)
p2 <- plotTrajectoryHeatmap(trajGSM, pal = paletteContinuous(set = "horizonExtra"))
trajGIM <- getTrajectory(ArchRProj = projHeme5, name = "LymphoidU", useMatrix = "GeneIntegrationMatrix", log2Norm = FALSE)
p3 <- plotTrajectoryHeatmap(trajGIM, pal = paletteContinuous(set = "blueYellow"))
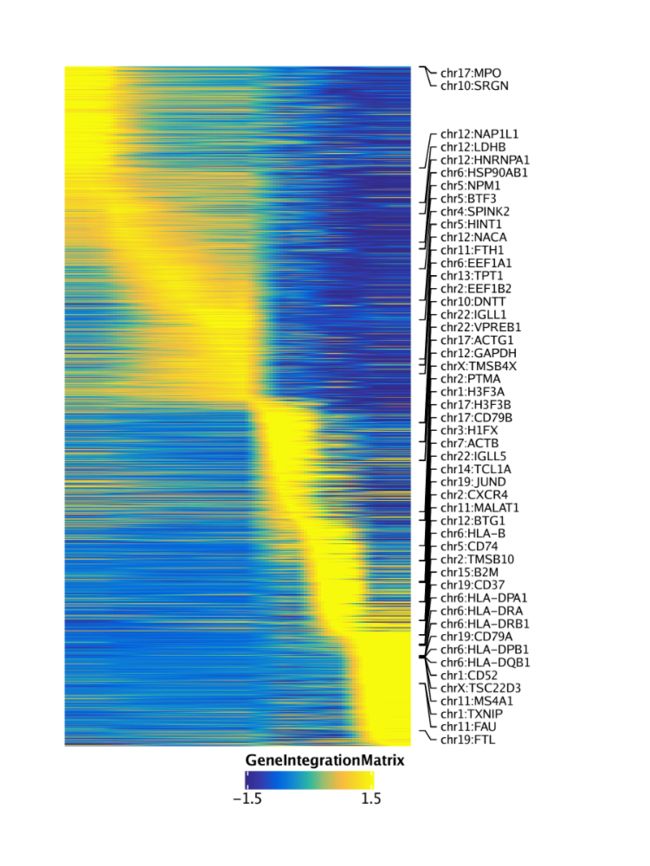
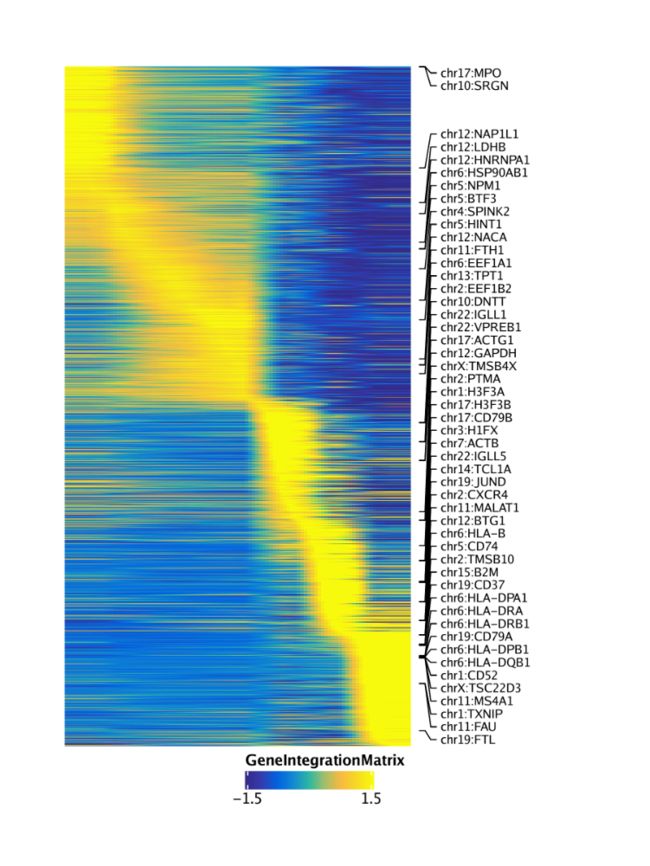
p4 <- plotTrajectoryHeatmap(trajPM, pal = paletteContinuous(set = "solarExtra"))
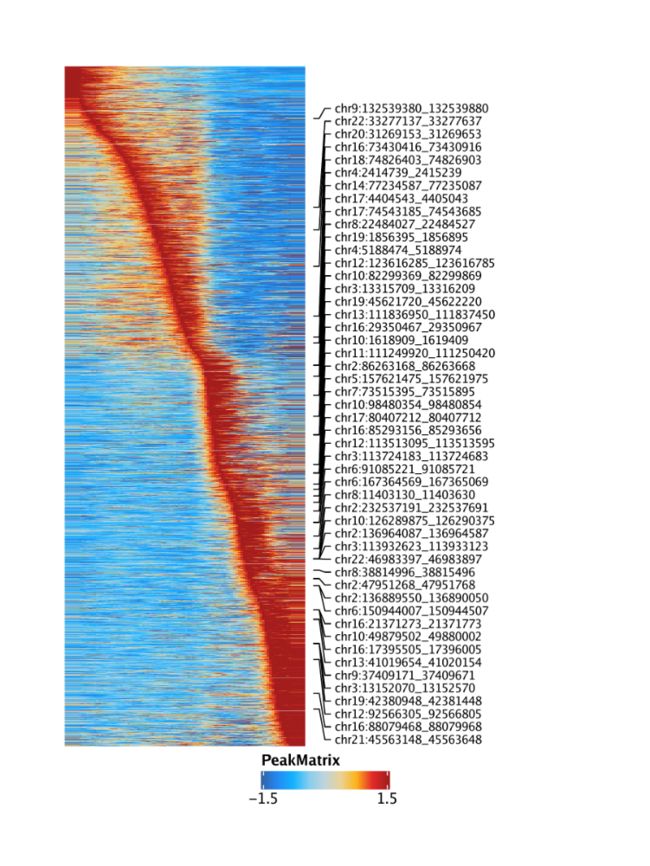
要保存此图的可编辑矢量化版本,请使用
plotPDF()。
plotPDF(p1, p2, p3, p4, name = "Plot-LymphoidU-Traj-Heatmaps.pdf", ArchRProj = projHeme5, addDOC = FALSE, width = 6, height = 8)
16.2.3综合伪时间分析
corGSM_MM <- correlateTrajectories(trajGSM, trajMM)
corGSM_MM[[1]]$matchname1
corGSM_MM[[1]]
trajGSM2 <- trajGSM[corGSM_MM[[1]]$name1, ]
trajMM2 <- trajMM[corGSM_MM[[1]]$name2, ]
trajCombined <- trajGSM2
assay(trajCombined) <- t(apply(assay(trajGSM2), 1, scale)) + t(apply(assay(trajMM2), 1, scale))
combinedMat <- plotTrajectoryHeatmap(trajCombined, returnMat = TRUE, varCutOff = 0)
rowOrder <- match(rownames(combinedMat), rownames(trajGSM2))
ht1 <- plotTrajectoryHeatmap(trajGSM2, pal = paletteContinuous(set = "horizonExtra"), varCutOff = 0, rowOrder = rowOrder)
ht2 <- plotTrajectoryHeatmap(trajMM2, pal = paletteContinuous(set = "solarExtra"), varCutOff = 0, rowOrder = rowOrder)
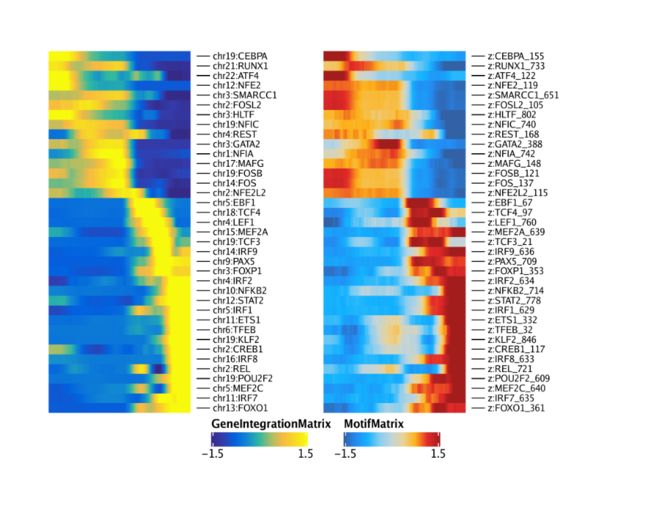
ht1 + ht2
corGIM_MM <- correlateTrajectories(trajGIM, trajMM)
corGIM_MM[[1]]$matchname1
corGIM_MM[[1]]
trajGIM2 <- trajGIM[corGIM_MM[[1]]$name1, ]
trajMM2 <- trajMM[corGIM_MM[[1]]$name2, ]
trajCombined <- trajGIM2
assay(trajCombined) <- t(apply(assay(trajGIM2), 1, scale)) + t(apply(assay(trajMM2), 1, scale))
combinedMat <- plotTrajectoryHeatmap(trajCombined, returnMat = TRUE, varCutOff = 0)
rowOrder <- match(rownames(combinedMat), rownames(trajGIM2))
ht1 <- plotTrajectoryHeatmap(trajGIM2, pal = paletteContinuous(set = "blueYellow"), varCutOff = 0, rowOrder = rowOrder)
ht2 <- plotTrajectoryHeatmap(trajMM2, pal = paletteContinuous(set = "solarExtra"), varCutOff = 0, rowOrder = rowOrder)
ht1 + ht2
参考材料:
https://www.archrproject.com/