- tcga数据下载_使用R包下载TCGA全部癌症的somatic突变信息
weixin_39671964
tcga数据下载
半年前我就系统性的介绍过:TCGA的pan-caner资料大全(以后挖掘TCGA数据库就用它)还专门指出了癌症的somatic突变的maf文件问题:TCGA数据库maf突变资料官方大全但是最近收到学员的反应,TCGA的maf文件开始控制下载了,下面是提问详情:非常久之前,整个TCGA数据库的全部数据都是提供下载的,包括fastq,bam,vcf,但是呢,后来因为保护病人隐私,就只开放maf格式的s
- TCGA 数据分析实战 —— 突变及拷贝数分析
名本无名
生信数据库R数据分析实战数据分析网络数据挖掘
TCGA数据分析实战——突变及拷贝数分析文章目录TCGA数据分析实战——突变及拷贝数分析前言基因组分析数据预处理识别recurrentCNVrecurrentCNV基因注释基因组变异可视化OncoPrintcircosplot部分区域可视化前言在介绍完TCGAbiolinks的查询下载和数据分析功能之后,我们简单展示几个示例,来练练手,加深对这个包的理解和使用我们主要从基因组、转录组和表观组3个维
- 常见生信分析
请你喝好果汁641
生信基础知识数据库oracle
转:https://mp.weixin.qq.com/s/fe9aZsgb-2U_s9Nvm4ImkA内容概览文章主要分为以下几个部分:TCGA数据挖掘GEO数据库挖掘单细胞全流程数据分析空间转录组分析多组学整合分析影像组学分析医学临床数据库介绍孟德尔随机化分析1.TCGA数据挖掘常见挖掘方向基因表达差异分析(RNA-seq)目标:筛选癌症与正常组织间表达显著差异的基因(DEGs)。工具:edge
- 生信初学者教程(九):数据预处理
生信学习者1
生信论文手把手保姆教程r语言数据可视化数据分析机器学习
文章目录LIRI-JPLIRI-JP临床表型加载R包导入数据清洗临床数据清洗实验处理数据清洗样品信息数据输出结果LIRI-JP转录组加载R包导入数据数据清洗过滤基因输出结果TCGA-LIHCTCGA-LIHC临床表型加载R包导入数据数据清洗输出结果TCGA-LIHC转录组加载R包导入数据数据清洗过滤基因表达值转换成countabundance输出结GSE14520GSE14520临床表型加载R包导
- lasso模型交替方向matlab_TCGA系列学习笔记(7)建模及模型评价
weixin_39782752
微信公众号:生信小知识关注可了解更多的教程及单细胞知识。问题或建议,请公众号留言;TCGA系列学习笔记(7)建模及模型评价内容目录前言1.背景知识1.1Cox前提假设的验证1.2lasso和ridge回归1.3C-index1.3.1如何计算1.3.2关于C-index的取值1.3.3用R计算C-index1.4Cox模型验证1.4.1数据拆分1.4.2ROC和AUC1.5逐步回归1.6nomog
- R语言 关联TCGA数据库下载的RNA-SEQ数据和临床信息
胖胖的胖球
生物信息学大数据生物信息学r语言
刚开始学习TCGA数据处理和分析,记下来方便以后查看setwd("E:/MyData/luadRNA-SEQ-20201028")#把工作目录定位到manifest文件所在的位置manifest="gdc_manifest.2020-10-28.txt"x=read.table(manifest,header=T)#header为TRUE表示读取第一行作为变量名表格已经建好了,可以view(x),
- 纯生信很难发表?只是你没有及时抓住研究热点
SCI狂人团队
当你还做meta分析的时候,你会发现meta分析很难发或者单位已经不承认了,而聪明的人已经开始做常规的生信GEO、TCGA数据挖掘这些(这个时候生信比较好发)。当你开始做常规的生信GEO、TCGA数据挖掘的时候,你会发现这些一样也是比较难发了,而聪明的人已经开始抓免疫评分这个热点进行生信数据挖掘(这个时候免疫评分比较好发)。当你开始对免疫评分这个热点进行生信数据挖掘的时候,你会发现自己的研究方向差
- 在TCGA上下载数据并且进行处理
Red Red
生信小技巧r语言数据库
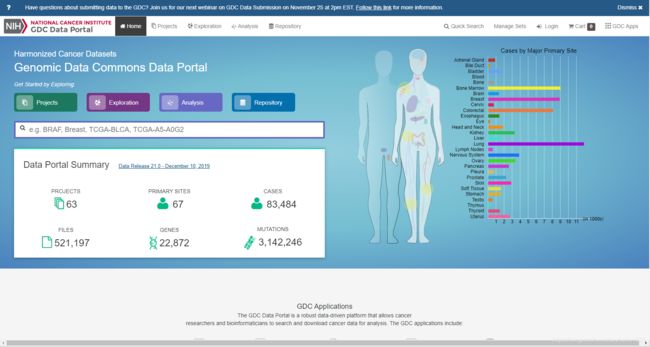
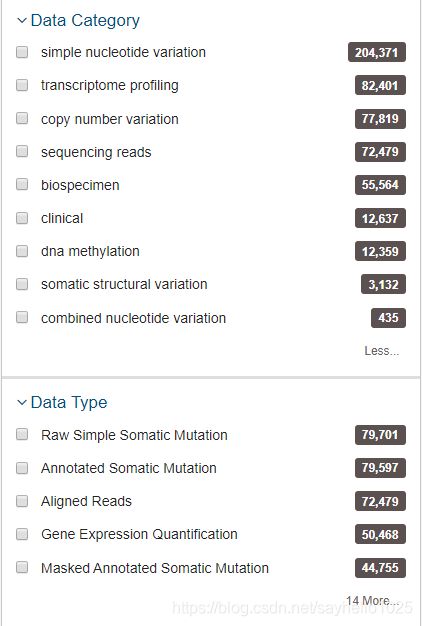
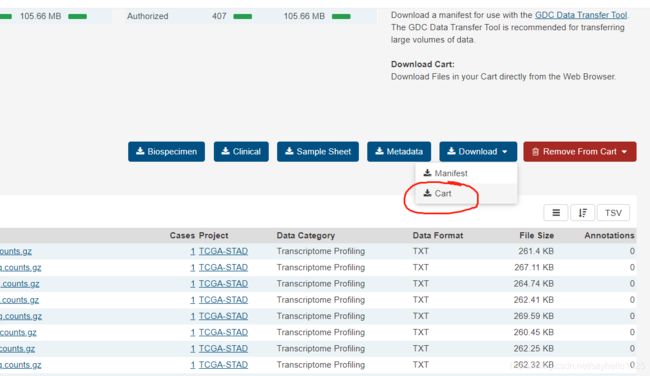
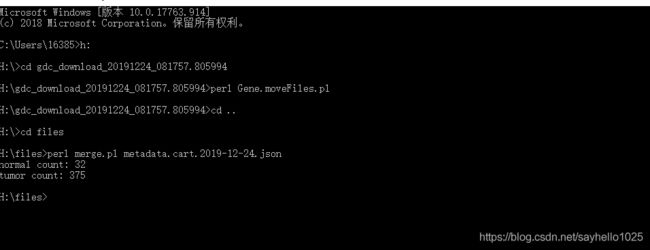
浏览器搜索TCGAGDC进入网站在TCGA数据库主页选择“Repository”模式根据所需要的选项在侧边栏选择数据清空购物车!!第一次登陆可忽略将刚刚选择好的数据加入购物车,并且在购物车里下载Metadata和Cart数据,下载到同一个文件夹下。使用R语言脚本对数据进行处理,将其提取为genesymbol和样本的数据,推荐看一下该博主处理数据!!真的非常详细!他R语言脚本在这个链接里
- 【TCGA】批量下载WSIs(GDC Transfer Tool-UI)
LansinBlog
TCGApython
从TCGA中批量下载WSIs1.下载TCGA原生工具TCGA官网链接:https://portal.gdc.cancer.gov/由于目前TCGA改版了,如果不太适应新版本界面的,可以点击红色部分,回到第一版。下滑找到“DataTransferTool”并点击。进入"DataTransferTool"子页面后,下滑找到“GDCTransferToolpage”并点击。进入“GDCTransferT
- 【R语言】批量读取某路径下文件内容
巩翔宇Ibrahimovic
R刚入门的时候,能够正确读取单个文件就觉得小有成就,随着时间的积累,单一文件地读取已经不能满足需求了,此时,批量地做就是解放双手地过程。使用for循环把下载地TCGA数据读入R语言并转换成数据框使用三个for循环来完成,这是第一个for循环。1.把所有数据读入在一个文件夹中dir.create("data_in_one")#创建目标文件夹,也可右键创建dir("rawdata/")#查看原
- 宝藏R包:TCGA的转录组数据挖掘一站搞定
小洁忘了怎么分身
最近在看ceRNA的时候看到了一个宝藏R包,写包简化了芯片数据下游分析之后,我正想着写转录组下游分析的简化版,就看到了它。用起来~0.R包和数据准备if(!require(GDCRNATools))BiocManager::install("GDCRNATools")library(GDCRNATools)这里使用的是作者给的示例数据,RNA-seq是1000行,miRNAseq是2588个。#m
- 一个实用的利用TCGA数据的网页工具
刘大帅1
如果你不懂代码,不懂网站规则,那么最简单的就是UCSC+xena+浏览器啦!!!https://xenabrowser.net/全部根据癌症种类整理好了,直接点击链接即可下载。同时也提供相应的可视化工具点击进入任何一个癌症数据:数据排列的整整齐齐,进入就是直接的下载链接。里面的数据集不仅仅包括tcga,还有其他许多的数据集。不懂的话可以看视频教学https://www.bilibili.com/v
- TCGA+biomarker——C-index
Clariom
通常情况下,通过以下几种指标来对模型进行评价。1)区分度:采用指标C-index和ROC曲线来评价区分度,一般文章都是二选一。C-index即一致性指数(indexofconcordance),通过评估模型预测结果与实际观察结果的符合程度,以评价模型的预测准确性。ROC曲线,展示特异性和敏感性,ROC曲线下的面积被称为AUC,它介于0.5和1之间,作为数值可以直观的评价分类器的好坏,值越大越好。2
- TCGA新版数据库表达矩阵提取
医学和生信笔记
本文首发于公众号:医学和生信笔记医学和生信笔记,专注R语言在临床医学中的使用,R语言数据分析和可视化。主要分享R语言做医学统计学、meta分析、网络药理学、临床预测模型、机器学习、生物信息学等。现在使用TCGAbiolinks下载转录组数据后,直接是一个SummarizedExperiment对象,这个对象非常重要且好用。因为里面直接包含了表达矩阵、样本信息、基因信息,可以非常方便的通过内置函数直
- TCGA数据下载系列之二:RTCGA
白云梦_7
library(RTCGA)library(RTCGA.clinical)library(RTCGA.rnaseq)library(RTCGA.mRNA)library(RTCGA.mutations)all_TCGA_cancers=infoTCGA()DT::datatable(all_TCGA_cancers)#指定任意基因从任意癌症里面获取芯片表达数据(这里是MRNA)expr3.9&`x
- 基因名称的转换
生信小鹏
从TCGA获得的数据名称为ensembleID,这对于后续分析是不方便的,我们需要把ensembleID转换成genesymbol和geneID。可以使用以下几种方法进行转换。1.使用gtf文件进行转换1.1下载gtf文件登录http://asia.ensembl.org/index.htmlimage.pngimage.pngimage.pngimage.png1.2制作R可读的gtf文件R读取
- 突然发现基本都是临床医生、医学生在搞纯生信数据挖掘
SCI狂人团队
在2016年之前,你在PubMed上搜索meta分析这个关键词会发现大部分相关的文章都是来自国内***医院或者***医科大学;而在2016年之后,来自国内***医院或者***医科大学的meta分析类文章数量明显下降,而在PubMed上输入TCGA、GEObioinformatics这些关键词会发现越来越多来自国内***医院或者***医科大学的文章。从这些文章数量的变化可以看出,由于很多单位政策的改
- TCGA简写对照表
中医封大夫
刚接触TCGA时,一看到肿瘤简写名称,总是头大,于是将对照表记录下来,以便随时查阅。Cohort英文名称中文名称ACCAdrenocorticalcarcinoma肾上腺皮质癌BLCABladderUrothelialCarcinoma膀胱尿路上皮癌BRCABreastinvasivecarcinoma乳腺浸润癌CESCCervicasquamouscellcarcinomaandendocerv
- 2020-02-25 Day30 利用生物信息学分析鉴定乳腺癌的潜在关键基因和关键通路
卅衣
image.png背景:在乳腺癌中,尚不完全了解肿瘤发生的分子机制。迫切需要鉴定与乳腺癌发展和预后相关的基因,并阐明其潜在的分子机制。在本研究中,我们旨在通过公共数据集的生物信息学分析来鉴定乳腺癌中潜在的病原性和预后差异表达基因(DEG)。方法:使用来自基因表达综合(GEO)和癌症基因组图谱(TCGA)的四个数据集(GSE21422,GSE29431,GSE42568和GSE61304)进行生物信
- 别人能发的纯生信,你不一定可以发
SCI狂人团队
很多人都是喜欢模仿别人的纯生信文章,例如看到这样的一篇纯生信文章:TheCancerGenomeAtlas(TCGA)basedm6Amethylation-relatedgenespredictprognosisinhepatocellularcarcinoma,马上模仿这篇文章,然后进行发表。如果你是刚开始接触纯生信的,模仿参考范文进行练手,的确是一种不错的选择;如果你已经发过了纯生信SCI,
- TCGA-302-下载数据实战
小梦游仙境
官网数据存放中心:https://portal.gdc.cancer.gov/image-20190619115407536image-20190619115504685先下载caseimage-20190619115611503image-20190619120123661接下来下载file,现在感兴趣的是miRNAseq的表达量image-20190619120258569image-2019
- TCGA的ID转换可以一步到位了
小洁忘了怎么分身
0.背景知识TCGA或TCGA+GTEx的表达矩阵,行名都是ensambleid,因为TCGA数据的参考基因组版本是genecodeV22,xena重新分析的TCGA+GTEx数据参考基因组版本则是genecodeV23。代码复制太多次了,于是我写了一个函数,将ensambleid表达矩阵直接转换为symbol。仍然是tinyarray包,今天说的函数是新写的,到Github下载最新版本的包吧:h
- 继续学习TCGA-使用R包TCGA-biolinks下载数据
程凉皮儿
参考学习资料:还是小洁老师的TCGA-3.R包TCGA-biolinks下载数据下载安装包>BiocManager::install("TCGAbiolinks")Bioconductorversion3.10(BiocManager1.30.9),R3.6.1(2019-07-05)Installingpackage(s)'TCGAbiolinks'tryingURL'https://mirro
- 五、生存分析前的数据整理
白米饭睡不醒
生存分析只需要tumor数据,不要normal,将其去掉,新表达矩阵数据命名为exprSet;GTEX可得到正常样本做补充(xena将GTEX和TCGA数据对应起来了)clinical信息需要进一步整理,成为生存分析需要的格式,新临床信息数据命名为meta。由于不同癌症的临床信息表格组织形式不同,这里的代码需要根据实际情况修改。rm(list=ls())options(stringsAsFacto
- 2022新版TCGA批量下载表达矩阵及临床信息
科研小徐
#BiocManager::install("BioinformaticsFMRP/TCGAbiolinksGUI.data")#BiocManager::install("BioinformaticsFMRP/TCGAbiolinks")gdcdata=function(i){library(TCGAbiolinks)projects%as.data.frame()%>%select(proje
- TCGA合并GETx数据进行免疫lncRNA提取,代码提问
叶间露
问题如下:下载TCGA肿瘤样本,但该数据中无正常组织,合并GETx数据进行免疫lncRNA提取,代码部分如下:rm(list=ls())library(limma)corFilter=0.4pvalueFilter=0.001setwd("C:\\Users\\CXH\\Desktop\\120irlncRNA\\09.irRNA")#读取lncRNA,并对数据进行处理rt=read.table(
- TCGA 数据分析实战 —— 富集分析
名本无名
前言通常,在识别完了差异基因之后,都会对差异基因进行功能富集,来获取差异基因参与的潜在生物学功能通路或生物学进程,有助于理解基因之间的作用关系以及发现基因在癌症发生发展过程中发挥的作用。通路,通常是一些已知的功能相关的基因集合,而我们常说的基因集合,一般是忽略了基因之间互作关系的通路。最常见的通路富集,是使用GO和KEGG数据库中预定义的生物学通路。1.GeneOntology(GO)GeneOn
- 【R语言】临床特征分组,多分类转换成二分类
生信交流平台
前面我们讲过肿瘤TNM分期我们知道T分期一般可以分成T1,T2,T3和T4四个期。另外一个常用的临床特征是组织病理分期,一般也是分为四期stageI,stageII,stageIII和stageIV。四组在我们做差异表达分析的时候是比较麻烦的。☞R代码TCGA差异表达分析☞零代码TCGA差异表达分析最简单的方法是将四个期合并成两个期。今天天我们就来聊聊如何用R来将四分期的临床特征转换成二分期。首先
- 纯生信分析套路 甲基转移酶相关因子的预后分析
音十千寻
今天跟大家分享的是2020年4月底发表在FrontiersinOncology(IF:4.85)上的一篇文章,有套路(分型-筛marker-预后模型构建,简单吧),只是关注的点变成甲基化转移酶相关因子。甲基化是一种可逆修饰,调控这种修饰的蛋白质和基因可以作为肿瘤治疗的候选靶点。然而,在胶质瘤中甲基转移酶相关基因的特征尚不清楚。本文主要是基于TCGA数据和中国胶质瘤基因组图谱RNA测序数据集,通过一
- Survival curve for TCGA
osho_c837
cgdsrlibrary(cgdsr)library(DT)mycgds=CGDS("http://www.cbioportal.org/")test(mycgds)###allcancerstudiesall_tcga_studies%droplevels()time_status$PERSON_NEOPLASM_CANCER_STATUS%droplevels()time_status$PER
- TOMCAT在POST方法提交参数丢失问题
357029540
javatomcatjsp
摘自http://my.oschina.net/luckyi/blog/213209
昨天在解决一个BUG时发现一个奇怪的问题,一个AJAX提交数据在之前都是木有问题的,突然提交出错影响其他处理流程。
检查时发现页面处理数据较多,起初以为是提交顺序不正确修改后发现不是由此问题引起。于是删除掉一部分数据进行提交,较少数据能够提交成功。
恢复较多数据后跟踪提交FORM DATA ,发现数
- 在MyEclipse中增加JSP模板 删除-2008-08-18
ljy325
jspxmlMyEclipse
在D:\Program Files\MyEclipse 6.0\myeclipse\eclipse\plugins\com.genuitec.eclipse.wizards_6.0.1.zmyeclipse601200710\templates\jsp 目录下找到Jsp.vtl,复制一份,重命名为jsp2.vtl,然后把里面的内容修改为自己想要的格式,保存。
然后在 D:\Progr
- JavaScript常用验证脚本总结
eksliang
JavaScriptjavaScript表单验证
转载请出自出处:http://eksliang.iteye.com/blog/2098985
下面这些验证脚本,是我在这几年开发中的总结,今天把他放出来,也算是一种分享吧,现在在我的项目中也在用!包括日期验证、比较,非空验证、身份证验证、数值验证、Email验证、电话验证等等...!
&nb
- 微软BI(4)
18289753290
微软BI SSIS
1)
Q:查看ssis里面某个控件输出的结果:
A MessageBox.Show(Dts.Variables["v_lastTimestamp"].Value.ToString());
这是我们在包里面定义的变量
2):在关联目的端表的时候如果是一对多的关系,一定要选择唯一的那个键作为关联字段。
3)
Q:ssis里面如果将多个数据源的数据插入目的端一
- 定时对大数据量的表进行分表对数据备份
酷的飞上天空
大数据量
工作中遇到数据库中一个表的数据量比较大,属于日志表。正常情况下是不会有查询操作的,但如果不进行分表数据太多,执行一条简单sql语句要等好几分钟。。
分表工具:linux的shell + mysql自身提供的管理命令
原理:使用一个和原表数据结构一样的表,替换原表。
linux shell内容如下:
=======================开始
- 本质的描述与因材施教
永夜-极光
感想随笔
不管碰到什么事,我都下意识的想去探索本质,找寻一个最形象的描述方式。
我坚信,世界上对一件事物的描述和解释,肯定有一种最形象,最贴近本质,最容易让人理解
&
- 很迷茫。。。
随便小屋
随笔
小弟我今年研一,也是从事的咱们现在最流行的专业(计算机)。本科三流学校,为了能有个更好的跳板,进入了考研大军,非常有幸能进入研究生的行业(具体学校就不说了,怕把学校的名誉给损了)。
先说一下自身的条件,本科专业软件工程。主要学习就是软件开发,几乎和计算机没有什么区别。因为学校本身三流,也就是让老师带着学生学点东西,然后让学生毕业就行了。对专业性的东西了解的非常浅。就那学的语言来说
- 23种设计模式的意图和适用范围
aijuans
设计模式
Factory Method 意图 定义一个用于创建对象的接口,让子类决定实例化哪一个类。Factory Method 使一个类的实例化延迟到其子类。 适用性 当一个类不知道它所必须创建的对象的类的时候。 当一个类希望由它的子类来指定它所创建的对象的时候。 当类将创建对象的职责委托给多个帮助子类中的某一个,并且你希望将哪一个帮助子类是代理者这一信息局部化的时候。
Abstr
- Java中的synchronized和volatile
aoyouzi
javavolatilesynchronized
说到Java的线程同步问题肯定要说到两个关键字synchronized和volatile。说到这两个关键字,又要说道JVM的内存模型。JVM里内存分为main memory和working memory。 Main memory是所有线程共享的,working memory则是线程的工作内存,它保存有部分main memory变量的拷贝,对这些变量的更新直接发生在working memo
- js数组的操作和this关键字
百合不是茶
js数组操作this关键字
js数组的操作;
一:数组的创建:
1、数组的创建
var array = new Array(); //创建一个数组
var array = new Array([size]); //创建一个数组并指定长度,注意不是上限,是长度
var arrayObj = new Array([element0[, element1[, ...[, elementN]]]
- 别人的阿里面试感悟
bijian1013
面试分享工作感悟阿里面试
原文如下:http://greemranqq.iteye.com/blog/2007170
一直做企业系统,虽然也自己一直学习技术,但是感觉还是有所欠缺,准备花几个月的时间,把互联网的东西,以及一些基础更加的深入透析,结果这次比较意外,有点突然,下面分享一下感受吧!
&nb
- 淘宝的测试框架Itest
Bill_chen
springmaven框架单元测试JUnit
Itest测试框架是TaoBao测试部门开发的一套单元测试框架,以Junit4为核心,
集合DbUnit、Unitils等主流测试框架,应该算是比较好用的了。
近期项目中用了下,有关itest的具体使用如下:
1.在Maven中引入itest框架:
<dependency>
<groupId>com.taobao.test</groupId&g
- 【Java多线程二】多路条件解决生产者消费者问题
bit1129
java多线程
package com.tom;
import java.util.LinkedList;
import java.util.Queue;
import java.util.concurrent.ThreadLocalRandom;
import java.util.concurrent.locks.Condition;
import java.util.concurrent.loc
- 汉字转拼音pinyin4j
白糖_
pinyin4j
以前在项目中遇到汉字转拼音的情况,于是在网上找到了pinyin4j这个工具包,非常有用,别的不说了,直接下代码:
import java.util.HashSet;
import java.util.Set;
import net.sourceforge.pinyin4j.PinyinHelper;
import net.sourceforge.pinyin
- org.hibernate.TransactionException: JDBC begin failed解决方案
bozch
ssh数据库异常DBCP
org.hibernate.TransactionException: JDBC begin failed: at org.hibernate.transaction.JDBCTransaction.begin(JDBCTransaction.java:68) at org.hibernate.impl.SessionImp
- java-并查集(Disjoint-set)-将多个集合合并成没有交集的集合
bylijinnan
java
import java.util.ArrayList;
import java.util.Arrays;
import java.util.HashMap;
import java.util.HashSet;
import java.util.Iterator;
import java.util.List;
import java.util.Map;
import java.ut
- Java PrintWriter打印乱码
chenbowen00
java
一个小程序读写文件,发现PrintWriter输出后文件存在乱码,解决办法主要统一输入输出流编码格式。
读文件:
BufferedReader
从字符输入流中读取文本,缓冲各个字符,从而提供字符、数组和行的高效读取。
可以指定缓冲区的大小,或者可使用默认的大小。大多数情况下,默认值就足够大了。
通常,Reader 所作的每个读取请求都会导致对基础字符或字节流进行相应的读取请求。因
- [天气与气候]极端气候环境
comsci
环境
如果空间环境出现异变...外星文明并未出现,而只是用某种气象武器对地球的气候系统进行攻击,并挑唆地球国家间的战争,经过一段时间的准备...最大限度的削弱地球文明的整体力量,然后再进行入侵......
那么地球上的国家应该做什么样的防备工作呢?
&n
- oracle order by与union一起使用的用法
daizj
UNIONoracleorder by
当使用union操作时,排序语句必须放在最后面才正确,如下:
只能在union的最后一个子查询中使用order by,而这个order by是针对整个unioning后的结果集的。So:
如果unoin的几个子查询列名不同,如
Sql代码
select supplier_id, supplier_name
from suppliers
UNI
- zeus持久层读写分离单元测试
deng520159
单元测试
本文是zeus读写分离单元测试,距离分库分表,只有一步了.上代码:
1.ZeusMasterSlaveTest.java
package com.dengliang.zeus.webdemo.test;
import java.util.ArrayList;
import java.util.List;
import org.junit.Assert;
import org.j
- Yii 截取字符串(UTF-8) 使用组件
dcj3sjt126com
yii
1.将Helper.php放进protected\components文件夹下。
2.调用方法:
Helper::truncate_utf8_string($content,20,false); //不显示省略号 Helper::truncate_utf8_string($content,20); //显示省略号
&n
- 安装memcache及php扩展
dcj3sjt126com
PHP
安装memcache tar zxvf memcache-2.2.5.tgz cd memcache-2.2.5/ /usr/local/php/bin/phpize (?) ./configure --with-php-confi
- JsonObject 处理日期
feifeilinlin521
javajsonJsonOjbectJsonArrayJSONException
写这边文章的初衷就是遇到了json在转换日期格式出现了异常 net.sf.json.JSONException: java.lang.reflect.InvocationTargetException 原因是当你用Map接收数据库返回了java.sql.Date 日期的数据进行json转换出的问题话不多说 直接上代码
&n
- Ehcache(06)——监听器
234390216
监听器listenerehcache
监听器
Ehcache中监听器有两种,监听CacheManager的CacheManagerEventListener和监听Cache的CacheEventListener。在Ehcache中,Listener是通过对应的监听器工厂来生产和发生作用的。下面我们将来介绍一下这两种类型的监听器。
- activiti 自带设计器中chrome 34版本不能打开bug的解决
jackyrong
Activiti
在acitivti modeler中,如果是chrome 34,则不能打开该设计器,其他浏览器可以,
经证实为bug,参考
http://forums.activiti.org/content/activiti-modeler-doesnt-work-chrome-v34
修改为,找到
oryx.debug.js
在最头部增加
if (!Document.
- 微信收货地址共享接口-终极解决
laotu5i0
微信开发
最近要接入微信的收货地址共享接口,总是不成功,折腾了好几天,实在没办法网上搜到的帖子也是骂声一片。我把我碰到并解决问题的过程分享出来,希望能给微信的接口文档起到一个辅助作用,让后面进来的开发者能快速的接入,而不需要像我们一样苦逼的浪费好几天,甚至一周的青春。各种羞辱、谩骂的话就不说了,本人还算文明。
如果你能搜到本贴,说明你已经碰到了各种 ed
- 关于人才
netkiller.github.com
工作面试招聘netkiller人才
关于人才
每个月我都会接到许多猎头的电话,有些猎头比较专业,但绝大多数在我看来与猎头二字还是有很大差距的。 与猎头接触多了,自然也了解了他们的工作,包括操作手法,总体上国内的猎头行业还处在初级阶段。
总结就是“盲目推荐,以量取胜”。
目前现状
许多从事人力资源工作的人,根本不懂得怎么找人才。处在人才找不到企业,企业找不到人才的尴尬处境。
企业招聘,通常是需要用人的部门提出招聘条件,由人
- 搭建 CentOS 6 服务器 - 目录
rensanning
centos
(1) 安装CentOS
ISO(desktop/minimal)、Cloud(AWS/阿里云)、Virtualization(VMWare、VirtualBox)
详细内容
(2) Linux常用命令
cd、ls、rm、chmod......
详细内容
(3) 初始环境设置
用户管理、网络设置、安全设置......
详细内容
(4) 常驻服务Daemon
- 【求助】mongoDB无法更新主键
toknowme
mongodb
Query query = new Query(); query.addCriteria(new Criteria("_id").is(o.getId())); &n
- jquery 页面滚动到底部自动加载插件集合
xp9802
jquery
很多社交网站都使用无限滚动的翻页技术来提高用户体验,当你页面滑到列表底部时候无需点击就自动加载更多的内容。下面为你推荐 10 个 jQuery 的无限滚动的插件:
1. jQuery ScrollPagination
jQuery ScrollPagination plugin 是一个 jQuery 实现的支持无限滚动加载数据的插件。
2. jQuery Screw
S