基于RNA测序技术的转录组从头拼接算法研究
基于RNA测序技术的转录组从头拼接算法研究
摘要:
生物信息学主要研究分子生物学领域,而对于分子生物学领域,转录组的从头拼接又是其核心内容,即利用转录组的测序片段拼接出整个转录组中的所有表达的转录体。而RNA测序的出现,在计算上给转录组的拼接提供了一定的挑战。在目前,转录组的拼接算法主要是基于参考基因组的拼接方法与从头拼接方法。虽然基于参考基因组的方法比从头拼接方法更有突破性,不过基于参考基因组的拼接方法,仍然存在着一定的致命缺点,即为要有一个高质量的参考基因组。而从实际情况分析,绝大多数的生物根本不存在一个可供参考的已知基因组,相比之下,头拼接算法的重要性就突显而出。基于该现象,本文主要在分析当前拼接算法的基础上,提出了一个全新的转录组从头拼接算法(Bridger),巧妙地利用基于参考基因组算法的一些技巧来弥补目前从头拼接算法的不足。借助人、狗与老鼠的RNA测序数据上的测试结果,来表明Bridger比当前所有的从头拼接算法突出。除此之外,还将通过例子展示了Bridger在实际应用中重要价值。最后,提出总结,进一步介绍了转录组拼接下游的一些研究工作与研究方向。
关键词:生物信息;参考基因组;拼接算法;测序数据
Abstract:
The main field of bioinformatics research in molecular biology, and for the field of molecular biology, scratch spliced transcripts group is the core content, namely the use of transcriptase sequencing fragments of mosaic in the entire transcriptase expression of all transcripts. The emergence of RNA sequencing, spliced transcripts in the calculation to provide a certain set of challenges. At the moment, stitching algorithm transcriptase mainly splicing method reference genome from scratch stitching method. Although the reference genome-based approach is more than de novo mosaic method breakthrough, but based on the reference genome splicing method, there are still some fatal flaw, that is to have a high-quality reference genome. From the analysis of the actual situation, the vast majority of organisms does not exist an alternative known reference genome, by contrast, the importance of the first stitching algorithm to highlight out. Based on this phenomenon, this paper based on the analysis of the current stitching algorithm, proposed a new de novo transcriptase stitching algorithm (Bridger), clever use of some techniques based on the reference genome de novo algorithm to supplement the current stitching algorithm. With people, test results RNA sequencing data on dogs and rats, to show all current Bridger prominent than stitching algorithm from scratch. In addition, by way of example will demonstrate the important value of Bridger in practical applications. Finally, summarize, and further describes some research work and research transcriptase downstream splicing.
Keywords: bioinformatics; reference genome; stitching algorithm; sequencing data
目 录
摘要…………………………………………………………………………………………………..ii
第一章 绪论
第一章 分子生物学的背景知识
1.1生物学基础…………………………………………………………3
1.2测序技术............................................................................................3
1.2.1测序技术的发展…………………………………………………………………………3
1.2.2单端测序和双端测序……………………………………………4
1.2.3 RNA 测序………………………………………………………………………………….5
1.3 测序片段的拼接……………………………………………………………………………6
第二章 拼接算法的研究现状分析
2.1基因组的拼接算法 ………………………………………………………………………14
2.1.1贪婪方法 ………………………………………………………………………………….16
2.1.2重叠-排列-共有序列的方法………………………………………………………17
2.1.3 De Bruijn 图方法………………………………………………………………………..18
2.2基于参考基因组的转录组拼接算法…………………………………………….20
2.3基因组从头拼接算法………………………………………………22
2.4转录组从头拼接算法的改进………………………………………25
第三章Bridger:新的转录组从头拼接算法
3.1拼接前的考虑………………………………………………………30
3.1.1测序数据的特点…………………………………………………30
3.1.2测序错误的预处理………………………………………………31
3.2算法的创新点………………………………………………………32
3.2.1舍弃deBruijn图而去构造剪接图……………………………32
3.2.3引进兼容图和最小路覆盖模型…………………………………37
3.2.4通过加权巧妙地利用测序深度信息……………………………38
3.3 Bridger算法………………………………………………………39
3.3.1利用RNA测序片段构造剪接图…………………………………40
3.3.2构造兼容图………………………………………………………41
3.3.3寻找最优的转录体集合…………………………………………43
第四章Bridger的测评
4.1测试数据和评价标准
…………………………………………………44
4.1.1测试的数据………………………………………………………45
4.1.2参数设置…………………………………………………………45
4.2评价方法和标准……………………………………………………47
4.3 Bridger的优缺点…………………………………………………49
4.4 Bridger的下游分析..……………………………………………………………………..51
第五章 总结
参考文献 ………….………………………………………………………………………………55
致谢词 …………………………………………………………………67
第一章 绪论
随着科技的增强,生物信息学的范畴也逐步增长,生物信息学是改革开放以来,出现的一门新颖的交叉学科,它主要的研究意义是利用数学、统计学以及信息学的相关知识,再结合计算机科学的相关研究知识去探讨并解决生物学上出现的问题。又随着生物学上的基因组测序技术的突破、而分子结构的测定技术在互联网的普及下又迈入了另一层面。
据此,生物数据在科技的冲击下,沿直线上增,数以百计的生物学数据库如同雨后春算般迅速成长,这些数据给生物学的研宄提供了一定的屏障。对于复杂多变的生物数据,生物学家们需要借助一定的数学算法和计算工具去研究并处理分析这些数据。然后在从生物数据中获取更多的有用信息,周而复始,逐步沉淀,从而形成生物信息学。
在当下,对于生物信息学的研究,则主要是集中在分子生物学这一领域上。而所谓的分子生物学,它主要是研究细胞内的核酸、脂肪、蛋白质等大分子的组成、形态、结构特征等等,根据这些特征的研究数据,人类可以真正地从分子平面上了解生物的特征与生活习性,进一步了解一些微生物的结构组成,在研究细胞内大分子的结构特征时,主要是研宄细胞内的DNA重组、遗传物质的表达、以及一些大分子的结构与功能等等。
经过几十年的研究,生物信息学的发展与突破就如同催化剂般在逻辑层次上极大限度地推动了分子生物学的进步与发展。与此同时,生物信息学在其他领域范畴内也产生了共鸣。在数学领域上、计算机科学领域内都留下了诸多具有挑战意味的学术问题。例如对于遗传物质的剪接研究,也有一定的冲击效果,本文也主要研究遗传物质的剪接问题。众所周知,在遗传物质的剪接过程中,mRNA(信使RNA)的产生并不单一,有着两种或者多种以上的mRNA,故此,通过翻译过程皆能产生两种或者多种以上的蛋白质,该现象在分子生物学上人们称其为可变剪接。
对于可变剪接,在生物学中,若真核生物经过一定的手段处理,基因中的内含子就会被处理掉,而所有的外显子就会相连。而基因中的外显子之间就获得不同的组合方式,从而形成不种种类的信使RNA。
就目前而言,在信息生物学上,可变剪接的方法常常用于对真核生物的处理,且越来越普遍,对于人类而言,基因中都具有多个外显子,这也是说,在人类的细胞内大约有95%以上的基因都存在着可变剪接。可变剪接的出现极大程度地增加编码的种类,这也是体现蛋白质多样性的主要因素。
在对细胞结构的研究中,我们已然知晓,细胞内所有的染色体物质皆构成一个庞大的基因组。而相对于真核生物的基因而言,原核生物的基因结构较于简普,简言之,原核生物的基因就是一段具有编码蛋白质的碱基序列,它并存在外显子与内含子,所以结构较为简单。真核生物的基因结构比原核生物的基因结构复杂得多,它不仅包括具有编码蛋白质功能的外显子(exon),还含有不具有编码蛋白质能力内含子(intron),外显子与内含子的存在也是分辨原核生物与真核生物的一大因素。
基因传递遗传信息是以蛋白质的形式表达,在表达过程中,蛋白质的形成包括两个阶段,其一为转录(transcription),其二则为翻译(translation)。转录是机体的遗传信息由DNA转向RNA的过程,即以双链DNA中的一条单链为模板,以四种核苷酸为原料,在RNA聚合酶的催化作用下生成RNA的过程,其中,四种核苷酸分别为腺嘌呤(A)、鸟漂呤(G)、胞啼唼(C)、尿喷唆(U)。在转录过程中,对于每一个RNA分子,我们都称之为转录体(transcript),而一个细胞又包含着成千上万的转录体,转录体的总和即为一个转录组。转录完成之后,才完成蛋白质合成的前一部分工作,转录体还需要进入下一环节,才能合成具有生物功能的蛋白质,此环节则为翻译,所谓翻译,也就是利用RNA合成蛋白质的过程。
在本文中,研究的主要问题主要针对于转录阶段,对于翻译阶段,且不坐谈。故此,我们主要着重于介绍蛋白质合成中的转录过程,翻译过程在此且不着分析。
对于基因的转录过程,一般分为预启动、启动、核糖核酸聚合酶与启动子脱离、转录延续与转录终止这五个阶段。在转录过程中,其主要是由DNA的一条链着手,借助于RNA聚合酶与其它转录蛋白的帮助,从而合成一条对应的核糖核酸序列。该序列的作用,就是将遗传基因中的存储信息完整地复制出来。在DNA转录成RNA时,我们通过等价关系,可以将DNA中的T转换成了RNA中的U,这样便简化分析。
以上言论,只对于真核生物,原核生物的转录阶段相对于真核生物,较为简单,其转录后的产物皆可以直接用于翻译蛋白质(个别噬菌体除外)。而真核生物的基因由编码蛋白质的外显子和不编码蛋白质的内含子组成,转录过程中,首先会形成mRNA前体,然后经过加工,再翻译成蛋白质。
转录组是合成蛋白质的核心部件,基于其重要性,本文则以当前一些转录组拼接算法为基础,去研究并提出了一个全新的转录组从头拼接算法(Bridger),巧妙地利用基于参考基因组算法的一些技巧来弥补目前从头拼接算法的不足。
第一章 分子生物学的背景知识
1.1生物学基础
众所周知,对于生物而言,其遗传密码是以DNA的形式存在于基因组中。而DNA分子是一个依照碱基互补配对原则,由两条核苷酸链构成的双螺旋结构的分子化合物。
在RNA的剪接过程中,由于RNA的剪接变化,转录体就会发生多种异常情况,而转录体的异常剪接,正是疾病产生的导火索。
就中国科学院近几年的研宄表明,生物所有的致病变异,就有三分之二的疾病变异产生于可变剪接阶段。在对癌症的研究中,异常的可变剪接时常发生在癌细胞中,这也是导致癌细胞增值的主要原因,是产生癌症的因素之一。因此,在对遗传物质的研究时,研究可变剪接过程,对于治疗许多人类疾病(包括癌症)都有着不可磨灭的意义。
1.2测序技术
测序技术的发展对于可变剪接的研究给予了一定的便利条件。为此,在研究可变剪接时,我们有必要了解第二代测序技术与RNA测序。掌握这些知识,在分子生物学中,对于分析测序数据与设计转录组拼接算法都异常常重要。
1.2.1测序技术的发展
20世纪50年代初,测序技术在生物界中横空出世,在测序技术出现的早期,人们就懂得利用化学降解的方法去研究并测定了DNA中的多聚核糖核苷酸序列。在后来,学者Sanger与Gilbert在此基础上又分别探寻出两种不同的DNA测序方法,这些测序方法的产生,有着及其重要的意义,它标志着第一代测序技术的诞生。而Sanger的双脱氧核苗酸末端终止测序法最终占据了第一代测序的主流地位。
又历经十余年,经过六个国家的不懈努力,在2001年,完成了人类基因组计划(Human Genome Project, HGP)。时光不老,科学家们经过十年的艰辛,终于在解读基因密码上取得了一定的突破,继而推出第二代测序技术。人类基因组计划的顺利完成,使我国生物学跨上了另一层面,也充分地证明了我国具有研究遗传信息的能力。
第二次测序技术的出现,是以Roche公司的454技术与Solexa技术和ABI公司推出的SOLiD技术为代表,对于第二代测序技术而言,它不仅饱含第一代测序技术的高准确性,还极大限度地提高了遗传信息的测序通量,测序通量的提高,标志着测序时间与测序成本得以降低。例如,在研究人类基因组计划时,科学家们就使用第一代测序技术作为研究手段,在此期间,花费了近 30亿美金的研究经费,历时十三余年;相比之下,如果利用第二代的SOLiD测序技术去研究一个人的全基因组测序,只需花费几千美金与一周的时间就可以完成这一项目。虽然说第二代测序技术具有很多优点,不过针对于第二代测序的测序数据分析,却给科学家们带来了一些问题与挑战,其原因在于第二代测序技术的测序片段较短、数据量大、不利于研究与分析。
在第二代测序技术的基础上,Helicos公司又推出单分子测序技术;Pacific Biosciences公司推出单分子实时测序技术;Oxford Nanopore Technologies公司的单分子测序技术,这些技术又统称为第三代测序技术,不过,由于诸多因素的影响,就目前而言,第三代测序技术并不成熟,错误率较高,很少使用于实际应用之中。由此,第二代测序技术仍然占据测序的主流地位。
1.2.2单端测序和双端测序
第二代测序技术Roche 454,Illumina和ABI SOLID有单端测序(singleend)和双端测序(pairedend)两种方式。在目前,这三种技术是诸多测序方式的三大主流技术。其中,对于Roche454测序技术来说,它主要是单端测序,它的测序片段长度可以达到400bp~800bp,在单端测序中,我们经常利用到基因组骨架的组装;而Solexa测序技术与ABISOLID测序技术的测序长度相对较短,其用途主要是用于对遗传信息基因组骨架的进一步组装与填补。不过Solexa技术与ABISOLID技术不仅可以单端测序,还可以双端测序。因此,只要合理运用双端测序的信息,就可以有效地克服测序片段短的缺点。
在本节中,我们主要以Illumina为例去介绍单端测序与双端测序。对于单端测序,其测序步骤大体总结如下:首先利用一定的方法,将DNA样本进行片段化处理,使其被打碎,从而形成200-500bp长度的测序片段。此步骤完成之后,又将引物序列连接到DNA片段的另一端,然后在引物序列的末端加上一个接头,最终固定片段,这样便能从测序序列的一端读取DNA中的碱基信息。而双端测序,就是在构建待测序的DNA库时,在测序序列的两端接头上都加上测序引物结合位点,继而进行两轮测序,这样便能依靠测序序列的两端来获得DNA中的碱基信息。
在实际应用中,在设计不同数据的拼接算法时,首先我们一定要充分考虑到是否利用双端测序技术。如果利用,就应该注意以下两个方面,其一,对于DNA测序片段的本身序列信息分析一定要合理;其二,双端测序的配对信息应有效利用。这样,才能给拼接过程提供更为权威的指导与参考。
1.2.3 RNA 测序
在设计拼接算法时,我们有必要介绍一下RNA测序,对于RNA测序的定义,我们可以将其解释为将高通量的测序技术应用到mRNA(信使RNA)上,然后逆转录生成的cDNA,在此过程中,就会产生了RNA测序,对于产生的RNA测序,通常被称为RNA-seq。对于RNA测序的用途,据总结,主要有如下几个方面,第一,可以利用RNA测序去研究不同基因的mRNA种类与各自基因的表达量;第二,可以借助RNA测序去分析探讨基因不同样本间的表达差异,继而为转录组的研究开拓一个有效的研究手段。在RNA测序未出现之前,对于转录组问题的研宄,研究人员主要是利用基于杂交的基因芯片技术为主要研究工具,不过由于技术上的缺陷,基于杂交的基因芯片技术无法准确地检测到新的mRNA,除此之外,此技术的灵敏度有限,对于表达水平的微小变化,也不能检测出来。
在转录组的研究中,其技术主要有传统的基因芯片技术与EST技术,不过对于这两种技术而言,RNA测序具有诸多优点。例如,RNA测序可以达到单核苷酸的分辨率,能够确定出表达量很
低的转录体,还可以研究可变剪接等等,这些成果,以上两种技术只能望尘莫及。基于RNA测序的优越性,一些专家学者便以此预测,在今后的几年或者几十年内,RNA测序技术有可能会取代传统的基因芯片技术,成为转录组研究的主要工具。而在应用方面,随着时间的推移,RNA测序技术也被广泛地使用于转录组的研究。
RNA测序对于转录组的研究具有不可磨灭的划时代意义,不过凡事都具有两面性质,在生物信息学中,RNA测序技术产生的海量数据虽然给转录组的研究带来一定的机遇,不过在带来机遇的同时,也赋予了一定的挑战。因此,有效地对RNA测序数据进行合理的分析与探讨,是RNA测序技术是否在科学探索中获得一席之位的关键。关于RNA测序的主要目的,可以理解为它是研宄转录体的表达情况与比较多个不同信息样本间的转录组差异。其主要研究的问题包括了如何准确地确定DNA转录组中表达的转录体序列,在转录体序列得到表达之后,又如何估计基因中的转录体序列的表达量,继而依照转录体序列去探讨不同样本之间的差异性表达等等。在本章中,我们所谈论只是RNA测序研究的一个热点问题,即为从测序片段着手,去分析并拼接出基因中所含有的转录体序列,确定此序列,也是转录组研究的一大重要基石,在转录组的研究中必不可少,因此,合理掌握RNA测序,是研究生物信息学的基本要求之一。
1.3 测序片段的拼接
在生物学中,测序片段的拼接一直以来,都是生物界中最为棘手的问题之一,虽然诸多学者专家研究测序片段已经有了几十年的历程,不过至目前而言,这一领域的相关问题并未得到真正的解决。所谓测序拼接,其意义就是将基因测序所获得的短序列通过一些方法手段,从而还原成一个较为完整的序列,该过程则称之为测序拼接。
对于遗传物质DNA中的测序片段,在针对拼接此过程时,最后得到的是一个完整的基因组序列。然而拼接RNA测序片段,由于可变剪接的存在,在拼接中,往往会得到一些彼此之间不连通的图,且每一个图的含义都是代表着一个基因位置上的转录情况。在拼接过程中得到的每一个图,我们都可以利用一定的方法去寻找与之对应的全长的转录体路。也就是说,在对转录组的拼接时,最终所获得的产物并不是一个长的测序序列,而是不计其数的转录体序列,在生物学上,我们将全体转录体统称为一个转录组。
在研究转录组时,我们可以将其合理地比喻成以下这种情况,从理论上来说,如果我们将一本书复制多份,然后利用剪刀将这些书分别剪碎,形成千千万万个细小的碎片,在剪接的过程中,我们剪书的方式都不相同,最终在这些碎片之中,我们总会找到某一个碎片与其他碎片具有相同之处,也就是说,某一个碎片与其他碎片具有一定的重叠几率。而我们依靠碎片中,这些重叠的部分,最终是完全有可能将这本书的碎片重新组合,继而拼接成未剪接时的样子,依靠碎片重叠,将整个碎片组装成一本完整的书籍,这就是生物学中的序列拼接问题的最好举例。话虽如此,不过在解决实际情况时,我们所研究的问题,远远不止这些,也没有这么简单,因为事物具有一定的偶然性。就上述问题来说,假如在剪碎片时,由于自身的原因,我们将一千万个小碎片中的一部分碎片丢失了,而另外一部分碎片又不巧被一些东西污染,使其的外表发生了改变,那么这些碎片上的字迹就会被污染物遮挡,在研究中,就会大大地增加寻找碎片重叠的难度。在这种情况之下,我们是很难将这些碎片从新拼接出来的,我们在生活中遇见的拼接问题事实上与上述问题相似,情况变幻莫测,所以,对于某件事物,我们都应该从多层次的角度去探析这一问题。
在最初研究序列拼接的算法时,其性质也只是为了开发一些具有启发式的算法,然后通过寻找,最终获得与当前序列的一段具有最大的重叠区域的测序片段来不断地延长基因中的当前序列。频繁使用,最终这种方法逐渐演变成一种基于图的算法,该算法主要包括以下两种,第一种被人们称为:“重叠一一排列一一共有序列”的方法,而另外一种方法则是基于 de Bruijn 图的方法。
对于这两种方法的使用,它们都各自拥有着自己的优缺点,在第二代测序数据未出现之前,主要为第一代测序数据,在第一代测序数据的拼接之中,第一种方法“重叠一一排列一一共有序列”获得了成功的应用。由此可见,第一种方法的主流意义是先计算出基因中的测序片段的两两之间的重叠区域,然后再将这些测序片段重新,继而使其排列成一个新的图。每一个测序片段都来自于基因组中,而基因组序列应该穿过每一个测序片段,所以在研究时,我们应该从图中获得一个测序片段的路,利用这个测序片段的路最终求取最后的基因组序的所有转录体的序列,在求取中,也要对转录组进行一定的拼接。
在测序研究的初期,人们对于转录拼接的研究并未引起重视,他们只是简单地按照基因组的一些传统方法去拼接RNA测序的数据,不过却未得到理想的成果。追溯其原因,我们可以将其总结为:转录组的测序与拼接的本质都异于基因组。
虽然DNA测序的深度,在整个基因组上我们都可以将其看为相同,不过对RNA测序而言,它测序的深度并不相同,虽然RNA测序在不同的转录体之间或者在同一个转录体的不同位置都具有非常大的差异,在某些时候,其深度甚至相差了几个数量级。我们知道,基因组的拼接实质在于拼接出一个具有线性关系的基因组序列,不过基于转录组的拼接,又与基因组的拼接有着一定的差异。在转录组的拼接中,因为转录组中存在着可变剪接,所以每一个基因中,都有可能存在着很多个转录体,故而在转录组的拼接中就会形成数以万计的图,而这些图都能有效地刻画出一个基因的不同种剪接情况,此为两者不同的原因之一。又因为转录组的测序与基因组的测序有所差异,所以转录组的测序具有一定的链特异性。在拼接转录组时,我们就可以利用DNA中链的信息,从而拼接出具有重叠区域的转录体或者是具有重叠区域的反义转录体,而对于基因组拼接的算法,我们可以不用考虑其链中的特异性,也就没有几率获得转录体与反义转录体。由以上阐述,我们可以明确到转录组的拼接为基因组拼接拉开了挑战的序幕。
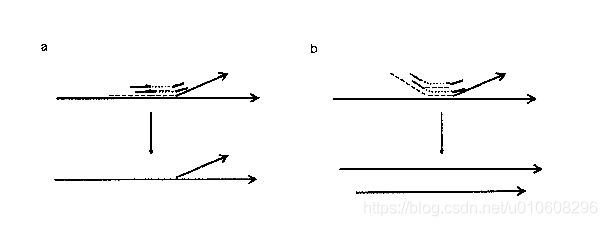
在逝去的几年时光内,对于这方面的知识,人们己经取得了不少的成果,且发明了许多转录组拼接算法。针对于这些转录组算法,我们可以根据它们是否需要一个参考基因组,将这些拼接算法分为如下两大类(如图1.1所示):

图1.1.基于参考基因组的转录组拼接和转录组从头拼接。
第一:基于参考基因组的拼接算法;第二:从头拼接算法。
首先,我们先来谈谈第一种算法,所谓的基于参考基因组的拼接算法,其步骤在于,先将需要测序的基因片段映射到基因组之上,这样不同的基因测序片段就会全部映射到基因组的不同位置,继而形成一个接着一个的测序片段簇,最终再通过一定的手段对这些测序片段簇进行拼接,这就是基于参考基因的拼接算法的大体步骤。而从头拼接算法的意义,从字面上我们已然知晓,从头拼接算法并不需要参考基因组,它是直接借助于RNA测序片段去拼接出最终我们所需的转录体,相对于参考基因的拼接算法,从头拼接算法的难度要大的多。
总体而言,基于参考基因组的拼接算法要比从头拼接算法简便,不过在简便的同时,基于参考基因组拼接却有一个较为明显的缺点,此缺点就是该算法必须依赖于一个参考基因组,而且对于参考基因组的质量要求也十分苛刻,换言之,基于参考基因拼接算法需要一个十分准确的参考基因组才能真正地获得最终转录体,不过在现实生活中,不是所有的物种都具有准确的参考基因,只有极少数物种的基因组达到这种要求,而绝大多数物种的基因组都是一个未知之谜,这给基于参考基因拼接算法带来了致命的打击。除此之外,有些物种就算存在高质量的参考基因组,这些参考基因组也不见得能使用于参考基因拼接算法中。
第二章 拼接算法的研究现状分析
在上节中,我们已经知道转录组的拼接与基因组的拼接有所差异,不过从算法上去研究该问题,两者之间又有着十分密切的关系。在本章节中,首先,我们先回顾基因组拼接中的常用算法。其次,再依次探究基于参考基因组的拼接算法与转录组从头拼接算法。最后,在前者的基础上,去总结并分析出当代生物学中关于转录组拼接研宄的现状与待解决的问题。
2.1基因组的拼接算法
对于基因组的拼接的概念,我们可以如此描述,从测序中所获取的短DNA序列片段出发,拼接出一个完整的基因组序列的过程就称之为基因组的拼接。在基因组拼接的问题刚刚萌芽之期,人们常常将其模型化处理,使其成为一个最短字符超串问题。而最短字符超串问题,其实质就是设定一个字符串集合为S,在集合S中找出一个最为短捷的字符串,使得集合中的字符串都是此字符串的子字符串,这便是最短字符超串问题。 以下,我们便举例说明该问题,下面是一个三个字符串和它对应的超串,如果将三个字符串中的每一个字符串都看成一个点,那么有重叠区域的两个字符串之间就会连成一条边,这样由点就可以构造出一个图。图构造出来之后,那么对于此问题,就可以将其演变成在一个图中寻找哈密尔顿路,所谓寻找哈密尔顿路,其实质是遍历图中的所有点,而且图中的每个点都只能遍历一次。在生物学上,哈密尔顿路的问题是一个及其富有挑战意味的问题,也没有一个相对而言的解法。不过,在后来人们受到物种杂交测序的影响,寻找出了一个稍微简单的解决方法,此方法就是在对物种的基因组拼接时,基因上的每一个测序片段被会被拆分成相互之间只有k-1个重叠核苷酸长度为k个的短序列,而且每一个长度为k的短序列都称为一个k-mer;下面是一个序列及其对应的一系列k-mer的例子:
ATCGTCAGG
ATCG
TCGT
CGTC
GTCA
CAGG
依靠以上例子,我们可以将这些K-mer建立成一个deBruijn图,每一个k-mer是图中的一个点,如果两个k-mer中有k-1个核苷酸的重叠区域,就给这两个点之间连一条边。
依照以上方法,我们就可以将基因组拼接问题模型化处理,就可以将哈密尔顿问题转换为在deBruijn图中寻找的欧拉路问题。而欧拉路问题,就是一条访问图中每一条边一次并且仅访问一次的路。由于哈密尔顿问题与欧拉路的建模方法有所差异,所以对于基因的拼接问题就从一个寻找哈密尔顿路问题转化为寻找欧拉路问题,这样,求解难度就在原有的基础上下降了不少。 在过去的几十年内,对于基因组拼接算法我们已经开发出诸多算法,通过总结,我们可以将这些算法分为如下三种类型:第一,贪婪方法;第二,“重叠~排列~共有序列”方法;第三,则为图方法,针对这三种方法,我们将在下节一一介绍。
2.1.1贪婪方法
在研究基因组拼接的初期,对于基因组拼接,学者们的处理方法都是借助贪婪方法来进行基因组拼接的,且许多拼接工具都是利用这一理论思想。 在基因组拼接中,贪婪拼接方法的基本思路主要是:在测序时,将具有重叠区域的测序序列进行一定的延长,且每一次延长,我们都要选择与当前序列的一端较长的重叠区域并且此重叠区域的相似度尽可能高的一条序列,然后再通过合并这两个重叠区域相似度极高的序列去延长当前序列,从而达到基因组拼接的效果。 以上我们简单地介绍了贪婪方法的基本思路,接下来,我们将重点介绍贪婪方法的步骤,对于贪婪方法的步骤,具体总结如下: 第一.计算出基因中测序片段的所有两两联配(alignment); 第二.在测序片段中,获得两个具有最大的重叠区域的序列; 第三.合并这两个重叠区域最大序列,使之成为一个新的序列; 第四.重复步骤二和三,直到剩下的所有序列小于某一个数值或者没有重叠区域。从步骤上看,我们已然知晓,序列的两两联配是贪婪算法的重要支持点,也是生物信息学研究的基本问题之一。
2.1.2重叠-排列-共有序列的方法
在测序中,重叠排列共有序列的方法是第一代测序数据的拼接中最为常用的方法,同时也是最成功的算法之一。在测序中,很多拼接工具都利用重叠排列共有序列方法,由此可见,重叠排列共有序列方法在测序中具有较高的实用性,因此,我们有必要对此方法进行一定的介绍,据总结,重叠排序共有序列方法的步骤如下:
第一.计算重叠区域。在测序拼接中,首先要计算出所有测序片段两两之间的重叠区域,计算重叠区域的意义是为了构造出一个重叠图,而重叠图中的每一个点都代表着一个测序片段,当两个测序片段之间具有重叠区域时,则给他们之间连一条边。
第二.图排列阶段。重叠图构造成功后,我们便在重叠图中寻找一些能够代表原始DNA序列的路。在理想情况下,在重叠图中,我们都希望获得一条相对完整的基因组序列,这样我们便可以从重叠图寻找到一个哈密尔顿路。
第三.确定共有序列。所谓确定共有序列,其实质就是确定一个序列,使该确定序列包含步骤二中的每一条序列,也就是说,该共有序列是步骤二中所有序列的子序列。
从效果上分析,贪婪方法的效果要次于重叠排列共有序列的方法,这也是重叠排序共有序列方法为什么能在第一代测序片段拼接中受欢迎的原因之一。虽然重叠排序共有序列具有高效性,不过与贪婪方法一样,重叠排序共有序列方法要计算测序片段两两之间的联配,而测序片段之间的联配的计算量十分浩大,故此,在第二代测序的拼接中,此方法的应用相对于第一代测序拼接要少得多。
2.1.3 De Bruijn 图方法
在二十一世纪初期,Pavel与Pevzner等人通过相关的研究探讨,最终提出了一个新的基因组拼接方法,这个拼接方法与贪婪方法和重叠排序共有序列方法并不相同,因为此方法并不直接利用测序序列,而只是通过一定的手段,将基因中的测序片段打碎,使之成为具有重叠区域的一系列长短序列,而打碎的短序列都具有相同的长度,且长度都为A个核苷酸,在此,我们都称之为tmer。且每一个测序片段都依次被人为打碎,从而成为相互之间都具有k-l个核苷酸的重叠A:mer。
因此,我们可以利用测序数据中所包含的的A:mer,然后去构造出一个deBruijn图,在此图中,对于每一个tmer,我们都可以将其看成是一个点,而两个tmer之间如果正巧有h个核苷酸重叠区域,那么这两个tmer之间就可以连一条边。正因为两个tmer之间如果有h个核苷酸重复区域可以连一条边,那么在测序过程中,就可以避免一些测序错误,构造出来的图与直接用测序片段构造的图有异曲同工之妙。在deBrujin图中,基因组所对应的一条能通过所有边的路,在生物学上,我们都称其为欧拉路。
这样,就可以将基因组的拼接问题摇身一变,使其转化为在deBrujin图上寻找到一个欧拉路的问题,而在图中寻找欧拉路,我们可以利用多项式的解法去处理该问题。
在基因拼接过程中,基于图方法的设计,并不是针对于第二代测序的拼接,从实际意义上研究,de Brujin方法出现的时间要早于第二代测序,在其出现之前,此方法就己经存在了。不过,由于该算法中deBruijn图的规模与基因测序数据的规模没有一点联系,这两者之间仅仅只跟基因组的大小有关联,故此,该方法特别适用于像第二代测序技术的高通量数据拼接中,这也是为什么该方法在第二代测序的拼接中得到了广泛应用的最佳解释。重叠排序共有序列方法之所以在第一代测序中取得不菲的成绩,其主要因素在于它所构造的图的规模跟测序片段的数量多少有一定的关联性,而拥有高通量数据的第二代测序技术,也正因为其数据量相对较大,导致图的规模较,最终出现无法求解的尴尬现象。
然而正因为份图这一优点,导致基因拼接过程比重叠排共有序列方法高效,所以在后来的研究中,基于第二代测序的基因组拼接算法,人们都差不多采用deBrujin这一方法。
以上阐述的观点基本上适用于理想状态,而在实际的拼接过程中,我们最终所获得基因组序列并不是一条十分完整的基因组序列,受到其他因素的影响,在基因拼接中,我们所获得的序列是几条或者几十条、以至于更多条且序列与序列之间没有重叠区域的一些DNA序列。故此,要想获得完整的基因组序列,还需要借助一些方法或者手段对这些序列进行进一步加工处理,这样才能充分利用基因双端的测序信息,求解出序列的方向与序列与序列间的对应位置。除此之外,我们还可以通过一些实验去填补基因组中的缺陷,最终解决问题获得一条较为完整的基因组序列。
2.2基于参考基因组的转录组拼接算法
在对绝缘拼接的过程中,基于参考基因组的拼接,主要涉及如下三个步骤:第一步,RNA-seq的数据被映射到参考基因组上。在此过程中,我们利用的映射工具主要有以下几大类:其中包括TopHat , SpIiceMap,MapSplice以及GSNAP等,这些映射工具对剪接具有一定的敏感。所谓对剪接敏感,其意义主要是指能够处理含有剪接连接处的测序片段。基因中的转录体是由不同的外显子连接而成的,在拼接中,转录体中间的内含子会被剪掉,若将内含子的测序片段直接映射在基因组上,那么测序片段的前后两部分就会被映射两个不同的位置,当然普通的映射工具无法完成这一工作。
将数据映射到参考基因上时,我们可以针对不同的位置,可以建立不同的图。对于每一个图,只要遍历图中的路,我们就可以获取基因中的所有转录体。参考基因组的算法的步骤一为:进行序列映射;序列映射是将测序所获的序列重新定位在基因组上。把测序片段定位在基因组上后,来自不同基因的测序片段就会被映射到基因组的不同位置,形成一个个测序片段簇。转录组的拼接问题就转化为单独的转录体拼接问题。对于每一个图,只要遍历图中的路,就可以找到最终的转录体。
比较有代表性的参考基因组的拼接算法是Cufflinks和Scripture。Cufflinks算法对映射到每一个基因位置的测序片段上定义了兼容关系。利用这种兼容关系,我们可以建立一个重叠图(over lap graph),在重叠图上,每一个测序片段可以看作图中的点,如果两个测序片段兼容,我们就给他们连一条边。重叠图中的路表示基因的各个转录体。在Cufflinks算法中,人们应用了一个最小路模型在图中寻找对应转录体的路。
Scripture算法的主要思路为:构造一个有向图连通性图(connectivity graph),构图思路是将基因组中的每一个碱基看成一个点,在两个碱基之间加一条边,继而构成图。从构造的图中,寻找到所有超过某一个阈值的测序深度的路,作为最后的转录体集合。据总结,参考基因组的方法主要有以下几个优点:
一,通过映射,可以将一个大的拼接问题分解成多个小的拼接问题。每一个小的拼接问题之间相互独立,从而提高了计算效率。
二,对于表达量比较低的转录体,具有一定的敏感性,对于检测较低的未翻译区,基于参考基因组的拼接算法具有十分重要的意义。
三,由于基因组是已知的,转录体间的小的空白区域可以依靠基因组序列填充。这样,转录体中,没有被测序片段也有可能被正确拼接。以上是基于参考基因组的拼接算法的优点,对于其缺点总结如下:
第一,要依靠参考基因组。且拼接成果取决于的参考基因组的质量。因为生物界中,大部分生物基因组都不是很完善,寻找已知基因组十分困难。
第二,拼接成果与序列片段映射过程息息相关,在映射过程中,许多映射不到的基因组序列会被丢弃,从而造成信息丢失,这样拼接成果会大打折扣。
由以上言论中,我们已经简便地知道基于参考基因组的拼接算法的优点与缺点,故此,在实际情况中,一定要结合实际,才能使结果更加趋于完善。
2.3基因组从头拼接算法
不利用参考基因组,仅仅从测序所得的片段出发,通过一定的方法最终将转录组的全部的转录体序列拼接出来的方法,称之为基因组从头拼接算法。基于基因组的拼接算法,对于低等的真核生物的转录体拼接而言,异常简便,不过对于真核生物的转录组拼接,效果却不太如意。因此,在对转录组拼接时,我们结合实际,分析转录组的特点,这样才能得到最佳的成果。
在诸多基因组从头拼接算法中,Trinit算法是第一个专门针对转录组设计的拼接算法,同时也是老鼠们公认的最好的转录组从头拼接算法。
其算法思想是依靠测序片段构造出一个长序列,然后在根据构造出的长序列构造出连通分支,针对长序列构造的连通分支,都能构造出一个图,然后利用穷举的方法寻找到路。
Trinit算法的具体步骤是:
第一步.利用贪婪策略拼接出长序列。在该步骤中,首先要构建出一个哈希表,然后在表中记录下所有的测序数据以及出现hmer出现的次数。
第二步,依靠第一步的相关数据,从而建立出de Bruijn转录体图。
在第一步中,长序列并并不能直接反映出转录组的复杂性,不过他保持着一个完整图的全部信息。因此,我们可以通过一些方法,去建造出一些图,构图思路为,将所有的长序列聚类成不同种类的连通分支,使连通分支内的长序列之间至少存在一个核昔酸的重叠区域;其次,为每一个连通分支建立图;最终,将测序片段全部映射图中。
第三步,从构造的图中,寻找到相关的转录体的路。对于寻找转录体的路,我们可以按照如下方法:合并deBruijn图中连续且不分叉的ytmer,使其形成一个更长的序列。除去由测序错误导致的小分叉边,使图得到简化。
图得到简化之后,我们可以通过一些动态规划过程与遍历图中路的方法去确定被测序片段支持的转录体。然后再运用测序片段自身的一些信息与双端测序的相关信息,将图中路的组合数降低,继而一一列举。
就目前而言,所有的从头拼接算法都是基于deBruijn图,因此ytmer的长度十分重要。一般来讲,对于ytmer的长度,大的A值在高表达的数据或者序列长度较长的数据上表现较好。而小的A值与其相反,它只是在低表达的数据或者序列长度较短的数据上表现较好。基于这一特点,老鼠们经常使用许多不同的值对转录体进行拼接,最后再将这些不同的拼接结果逐一合并,这样才能得到更加完美的拼接效果。用这种方法拼接转录组的软件大致包括Rnnotator、Multiple-A:、Trans~ABySS与Oases_M。
虽然这种策略能得到较高的敏感性,不过却引进更多的假阳性转录体,故此,该策略并不是一个较为理想的策略。
在Trinity的算法中,我们可以根据不同的转录组数据与基因组数据,在拼接过程中,老鼠们地使用很多具有转录组数据的拼接技巧,这样,才能使拼接效果有了突破性的进展。虽然Trinity算法的拼接效果十分明显,不过它仍然存在着很多缺点与不足之处,这需要我们在今后的学习中,逐步总结。
2.4转录组从头拼接算法的改进
在本节中,针对于不同的RNA测序数据,我们选用了目前效果最佳的两个算法,Trinity算法与Oases算法,然后利用模拟数据的方法对其进行系统化分析。从分析的结果中,我们不难发现,对于简单转录组的拼接,这些算法的表现还可以,不过对于相对来说,较为复杂的转录组的拼接,该算法的效果却不是十分明显以至于很差。
对于Trinity算法与Oases算法,我们可以通过老鼠与乳酸菌的数据表现去探究。从结果上看,老鼠的数据是依靠工具BEERS生成,它含有40,000,00个双端测序的测序片段,而每一个测序片段的长度为75bp,而插入长度为200bp。
在获得老鼠的相关数据之后,我们又利用Perl程序使之生成乳酸菌的RNA测序数据。在测试中,要保持乳酸菌的数据的可比性,数据量以及测序片段长度和插入长度与老鼠的数据量、测序片段长度一致。
如果一个拼接出来的转录体能够完全覆盖一个参考转录体,并且插入删除不超过序列长度的1%,我们就认为这个参考转录体被完整拼接了出来。
如果拼接出来的转录体的50%以上的序列不能映射到任何参考转录体上,就将其确定为一个错误的拼接,或者将其定位为一个假阳性的转录体。然而,对于拼接的质量,在生物学上,并没有一个固定的标准,因此我们可以采取以下两个度量去衡量拼接的质量,这两个词为敏感性与假阳性。
Trinity和Oases在老鼠和乳酸菌上的拼接的结果如图2.1所示。从图中可以看出,虽然Trinity和Oases的表现有差异,但是结果一致表明,当前的拼接算法对于乳酸菌的拼接,敏感性很高,假阳性很低,效果比较好,对于老鼠的转录组的拼接,表现却差了好多,不仅敏感性低,而且假阳性也比较高。
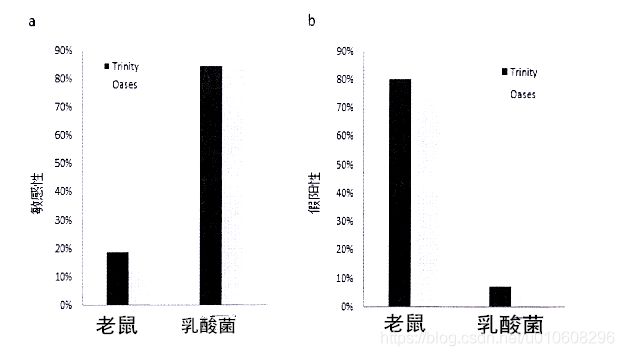
图2.1. Trinity和Oases在老鼠和乳酸菌上的拼接的结果
Trinity和Oases在五份数据的拼接结果如图2.2所示,随着转录组的复杂性的增加,拼接的敏感性迅速下降(图2.2a),假阳性也不断上升(图2.2b),进一步表明当前的拼接算法在处理复杂的转录组时表现很差。因此,非常有必要去进一步提高转录组的从头拼接算法。
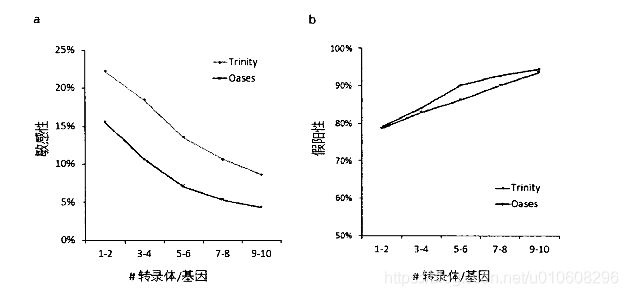
图2.2:测序数据的拼接结果
当转录组中的基因的剪接情况比较复杂时,拼接出来的图就相应的比较复杂,从这个图中去找对应转录体的路就变得很困难,所以,拼接的效果就变得往往很差了。因此,要想提高拼接的效果,必须在从图中寻找对应转录体的路这一步下功夫,建立一个更好的模型,充分利用各种信息,从而找到一个更好的路的集合。
我们应该提出的算法Bridget",我们舍弃了Trinity中利用的穷举算法,引入了一个经典的组合优化模型一一最小路覆盖模型,来找一个尽可能小的路的集合,来解释当前观察到的所有测序片段。仅仅这样还不能保证解的唯一性,我们又通过加权,通过优化来得到一组更好的解,加权后的模型在实际中几乎可以保证得到唯一解。
目前,老鼠们普遍将提高转录组从头拼接的质量寄希望于测序片段长度的增加。Mark Chaisson等人在研宄测序片段长度对于基因组的拼接的影响时,曾经给出过一个惊老鼠的结论:对于双端测序来说,测序片段的长度较短时,增加片段长度对拼接是很有用的,但是测序片段一旦超过某个阈值,增加测序片段的长度对拼接的结果影响不大。这个结论并不能不假思索地推广到转录组的拼接,毕竟转录组的拼接跟基因组的拼接有很大不同,那么对于转录组的拼接,是不是也有类似的结论呢?
为了弄清楚这个问题,我们通过模拟数据进行了深入研宄。首先,我们模拟了三个不同物种的RNA测序数据,老鼠,老鼠和乳酸菌。对于每一个物种,我们又分别生成了多份不同长度的数据。对于老鼠和老鼠,我们用一个模拟器BEERS分别生成了六份长度不同的RNA双端测序数据,序列长度分别为50bp, 75bp,lOObp, 150bp,175bp和200bp。每一份数据的中间插入的长度都为200bp,测序片段的数据量都是五千万左右,以保证有足够的测序深度。对于乳酸菌,我们利用一个Perl脚本生成了测序长度为35bp, 50bp, 75bp, lOObp和150bp的五份双端测序数据。对于每一份测序数据,中间插入的长度都是200bp,测序片段的数据量也都是五千万左右。

图2.3:老鼠RNA数据上的拼接结果比较
我们选用两个当前比较流行的拼接算法Trinity和Oases进行拼接,以防止因为某一个拼接算法自身的特点而导致错误的结论。对于拼接的结果,我们分别从敏感性和假阳性两个角度进行评价。虽然Trinity比Oases的拼接效果要好,但是随着测序长度的变化,二者的拼接效果的变化趋势是一致的。在老鼠的RNA测序数据上(图2.3),当测序片段长度从50bp增加到150bp时,两个算法的敏感性上升很快,敏感性也迅速下降,表明拼接的结果不断提高,但是当测序片段的长度超过150bp时,测序片段长度的继续增加对两个算法的拼接结果的影响微乎其微,具体表现为:敏感性的提高不到1%,假阳性几乎不再下降。在老鼠的测序数据上,也观察到了几乎一模一样的结果(图2.4)。

图2.4:乳酸菌RNA数据上的拼接结果比较
在乳酸菌的数据上,拼接的结果也是随着测序片段长度的增加表现出先有所提高后几乎没有什么变化的规律。不过有意思的是,在乳酸菌的数据上,测序片段的长度增加到75bp时,拼接的效果就基本达到了最优值,之后测序片段长度继续增加,对拼接几乎不再有任何影响。
可见,对于转录组双端测序的片段长度,也存在一个阈值(这个阈值对不同的物种是不同的),在这个阈值之下,测序片段的长度的增加对提高从头拼接的结果有一定的影响,但是,一旦超过了这个阈值,测序片段长度的增加对拼接结果就不再有任何影响。这一方面告诉我们,不能一味地寄希望测序技术的进步来提高转录组的从头拼接;另一方面,再一次告诉我们,要想从根本上来提高转录组的从头拼接,必须通过提高算法的途径来实现,因此对拼接算法的研宄和进一步改进十分必要。
第三章Bridger:新的转录组从头拼接算法
在本章中,我们主要介绍从头拼接算法Bridger。在实际测序数据时,Bridger算法的效果达到了我们的预期的设想,这大大地缩短了从头拼接算法与基于参考基因组的拼接算法之间的差异,这也是Bridger得名的原因。
3.1拼接前的考虑
3.1.1测序数据的特点
对于RNA测序数据的生成方式,主要为:将所有的RNA打碎,继而转化为一个c DNA文库,利用第二代测序技术对文库进行测序,从而产生n个测序片段,这里的n可以取任意值。在不同的测序平台上,测序的数据长度不同,测序数据的特点也出现多样性。
RNA测序 序的特点是:RNA测序可以做到具有链特异性(strand-specific)测序。该特点对于拼接是很重要的,特别是对于细菌或者低等的基因密集的真核生物而言,利用链的特异性信息会得到更佳效果。
DNA的两个不同的链上的转录体之间有重叠区域,在拼接时,如果不考虑链的信息,就很可能产生错误,误将两个转录体连接成一个更长的转录体,而利用链的信息,就可以有效地将两个转录体区分幵,从而正确地拼接出这两个转录体。对于高等生物的转录组来说,链的特异性的信息对于检测反义转录体也是很重要的。影响拼接的效果的另一个很重要的因素是测序的深度。对于转录组的从头拼接,如果测序深度过低,很多基因的表达可能检测不到,如果测序深度过高,会引进更多的测序错误,也影响拼接的结果。因此,一个合适的测序对于得到一个高质量的拼接至关重要。对于测序深度的研究没有引起人们足够的重视,研究相对比较少,直到最近,人们才对六种生物的转录组从头拼接所需要的测序深度进行了系统的研究。他们的研究发现,对于一个生物体的转录组从头拼接,大约需要三千万个测序片段,在这个测序深度下,拼接的效果可以达到一个比较好的状态;但是,当测序片段过多,比如超过六千万个时,由于数据中噪音过大,就会对拼接的效果产生负面的影响了。
3.1.2测序错误的预处理
测序错误是一个值得关注的问题,测序中的错误直接影响拼接的难度和拼接的最后结果。因此,一般在分析测序数据前,都要对测序数据进行预处理,对测序错误进行更正。事实证明,测序错误的更正能有效的提高拼接的效果。测序生成的数据中,每一个测序片段的每一个位置,都有一个测序质量的得分,一个较低的得分表示该位置发生测序错误的可能性比较大。在一个测序片段中,不同位置的测序错误率并不相同,通常是两端的错误率比较高,中间的测序错误率比较低。因此,人们得到测序数据后,通常先进行截尾处理,即去掉测序片段端点处错误率较高的几个碱基。另一种测序错误处理的方法就是利用yt-mer的频率来对测序测错进行更正。
其基本思想是:同一个转录体被测序了很多次,发生测序错误的位置往往是不同的,这样没有错误的&-mer通常会出现很多次;如果某个it-mer出现的频率很低,那么这个yt-mer中就极有可能是测序错误造成的。我们通常会去掉这些含有测序错误的片段或者yt-mer,以降低拼接过程中数据的噪音以及对内存的需要。目前,己经有很多测序错误的更正工具,很多拼接方法中也内置了这一步骤,这些工具主要包括ALLPATHS-LG,QUAKE,SGA,ECHO,PEPTILE[84]等。关于这些工具的比较,可参见最近的一项研究。
3.2算法的创新点
3.2.1舍弃deBruijn图而去构造剪接图
转录组拼接的一个很大的困难来自于基因的可变剪接,假设没有可变剪接,一个基因仅仅对应着一个转录体,那么拼接就变得简单多了。正是由于可变剪接的存在,让每一个基因的转录情况变得复杂,一个线性的序列不足以表示一个基因复杂的转录情况,而是需要用一个图结构来表示。当前所有的从头拼接算法都是利用RNA测序数据去构造deBruijn图,而在Bridger算法中,我们去构造一个能更直观地表示出一个基因的剪接复杂性的图线性序列来表示,如果一个基因有多个转录体,就表示成多条序列。而剪接图为此提供了一个很方便的表示,将一个基因表示成一个图,每一个外显子表示成图中的一个点,如果两个外显子在可变剪接的某个转录体中是前后直接相连的,则给他们之间加一条有向边,有向边的方向是从该基因5’端的外显子指向3’端的外显子。这样,一个基因的剪接图就是一个有向无圈图,基因的每一个转录体就对应着图中的一条路
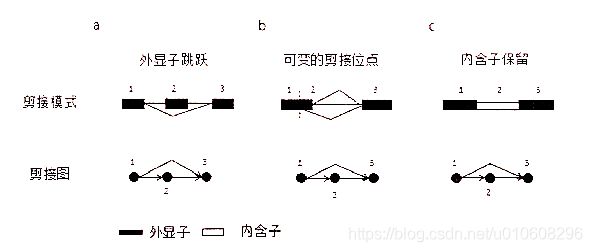
图3.1.几种可变剪接模式及其对应的剪接图。
- 外显子跳跃;b)可变的剪接位点;c)内含子保留。在本论文中,我们推广了剪接图的概念,主要是因为在拼接之前,基因的结构是未知的,很多时候无法准确地确定出一个外显子的边界。例如,某两个连续外显子在所有的转录体上都出现,始终连在一起,是根本无法区分两个外显子的边界的,在这种情况下,就把它们看作是一个外显子。3.2.2构造图的过程中利用了双端测序的信息
构造图的一个子步骤就是将一个短序列延长为一个长序列,通常的做法是找到跟当前序列末端的hmer有k-l个核苷酸重叠的一个A-mer,然后将当前序列延长一个核苷酸。例如,要将序列的3’端方向延长,就是找一个A:-mer,它的A:-1前缀跟当前序列3’端的最后一个hmer的k-l后缀完全E配,然后利用这个k-mer,将当前序列向3’端延长一个核苷酸(图3.2a)。
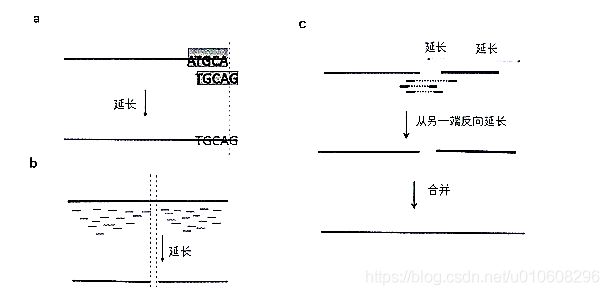
图3.2
当前所有的算法都是利用这种策略将一个序列向两端不断延长,直到不能继续延长为止,即找不到一个满足延长条件的A-mer。这样延长完之后,会产生如下问题:如果因为测序所得序列片段不能完全覆盖一个转录体(哪怕只有一个A-mer没被覆盖过来),就会导致一个转录体被拼接成了两部分。
如图3.3b所示,一个转录体序列被拼接成了两部分,但是这两部分本应该合并成一个更长的序列,才是一个正确的转录体。遗憾的是,在拼接的过程中,我们可能并不知道自己把一个序列拼成了两个短序列。即使有意识地去合并这两个短序列,最大的困难在于没有很好的办法将它们找出来,即不知道哪两个或者多个短序列原本来自于同一个转录体。其实,这个问题可以依靠双端测序的信息得到一定程度的解决。
在延长一个序列的时候,首先按照通常的延长策略去一步步延长,当不能延长时,将能够匹配到序列端点处的测序片段找出来,对于双端测序的数据,就可以找到这些测序片段对应的另一端的测序片段,利用这些另一端的测序片段可以进行反方向延长,延长得到同一个转录体的另一半序列,然后把这两个序列合并起来,从而得到完整的转录体序列。
通过利用双端测序的信息,很多短的序列被合并成一个长序列,使得拼接出的转录体更加完整。其实,我们之所以想到这个技巧,是受到基因组拼接过程的启发。在基因组拼接中,我们的目标是拼接出一个完整的基因组序列,但是实际中,往往是拼接出了几条甚至上百条长序列。在基因组拼接的后续处理中,就是通过双端测序的信息来确定这些长序列的前后位置和方向,然后把他们进一步合并起来。
最终,往往得到一个很大的图,很多基因混合到一起,给后面的找转录体提出了很大的挑战。在构造剪接图的时候,我们利用双端测序的信息进行检查,有效地控制了剪接图的大小,通常一个剪接图只有几个点到几十个点,在这么小的图中找出对应转录体的路,就变得简单多了。下面,我们通过一个有相同的yt-mer的两个基因的例子(图3.4)。
介绍如何利用双端测序信息来分开这两个基因,从而构造出两个较小的图,而不是一个将两个基因混在一起的较大的图。假设我们己经拼接出了其中的一个基因的序列,当发现某个yt-mer还可以继续延长时,就得到了图中的一个分支(如图3.4的红色实线)。
在不考虑测序错误的情况下,这个分支有两种可能:一种可能是该基因发生了可变剪接,另一种可能是某一个基因与该基因仅仅有一些相同的)t-mer。为了确定到底是哪一种可能性,可以利用双端测序的信息。首先找出这个分支靠近分支点部分的双端测序片段,然后找其另一端的测序片段,如果发现另一端的测序片段映射到了当前图(图3.4中蓝色实线)中,那么是第一种可能的概率就比较大,该分支就应该被加入到当前图中(图3.4a)。
如果发现另一端的测序片段不能映射到当前图中,则可以认定是后一种可能,这时该分支就不应该被加入到当前图中(图3.4b),而是在以后被单独地构造成另一个图,这拼接另一个图的时候,这些被两个基因共同拥有的hmer允许被再次使用(在构造deBruijn图中,每一个hmer只允许被使用一次),以保证在另一个图中可以形成完整的转录体,而不是一个转录体被分成两部分。图3.4.利用双端测序信息来控制剪接图的大小。找出分支上靠近分支点部分的双端测序片段,然后找其另一端的测序片段,当a)另一端的测序片段映射到当前图中时,将分支加入当前图;否则b)该分支不能加入当前图,但是该分支会被独立地构成另一个新的图。
3.2.3引进兼容图和最小路覆盖模型
不管是办图还是剪接图,一个转录体都对应着图中的一条路,但是并不是图中的每一个路都对应着一个真实的转录体。例如在图3.5a中,图中总共有四条路,但是实际可能只存在两个转录体(如图中对应紫色的路和绿色的路的转录体)。对于发生更加复杂可变剪接的基因,外显子之间的组合方式将会变得十分复杂,图中路的数目会呈现出指数性的增长。从图中寻找对应转录体的路,目前现有的方法基本是利用一些启发式的方法。例如,当前最好的转录组拼接算法Trinity中采用的是穷举法来寻找对应转录体的路。
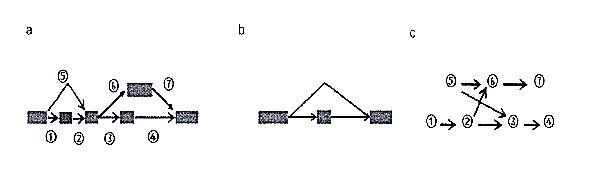
图3.5 构造兼容图
Trinity通过一个动态规划的过程,利用测序信息和一个打分机制来降低图中路的组合数,然后将这些序列一一穷举出来。这样预测出的转录体集合比较大,虽然有更大的可能性包含了正确的转录体,但是同时引进了很多假阳性转录体,这正是Trinity最大的缺陷。
3.2.4通过加权巧妙地利用测序深度信息
应用最小路覆盖模型,得到的解并不是唯一的,例如在图3.5c中不难观察至IJ,除了用绿色和紫色标识出来的两条路,还有两条路也可以覆盖图中所有的点,倘若得到那种结果,那就拼接错了。
为了确保得到正确的那个结果,我们考虑通过加权来实现。一个基因的不同转录体的表达是有差异的,表达的差异在测序上表现为测序的深度不同。来自同一个转录体不同部分的测序片段的深度是比较相似的。表达量高的跟表达量高的组合到一起,表达量低的跟表达量低的组合到一起,这样就保证了给出的路的集合以更大的概率得到正确的解。
3.3 Bridger算法
在综合分析现有算法的基础上,我们设计了一个全新的转录组拼接算法,命名为Bridger。Bridger首先由RNA测序数据出发,为数据中表达出来的所有基因建立剪接图。理想情况下,一个剪接图对应着一个基因的剪接情况。但是,由于存在同源基因和一些低表达的基因的影响,实际并非如此。但是,即使有时候将几个基因混在了一起,我们的方法仍然是有效的。构造完剪接图之后,Bridger利用剪接图再去构造兼容图,在兼容图上用一个严格的数学模型一一最小路覆盖模型来寻找一个路的集合,这个路的集合很容易转化为剪接图上路的集合,从而得到最终的转录体。图3.6中给出了Bridger算法的框架图。

图3.6.Bridger算法的流程图
3.3.1利用RNA测序片段构造剪接图
首先,利用测序片段构造一个哈希表,对于测序片段中出现的每一个/fc-mer,在哈希表键建立一个“键值”对。键即为该hmer的序列,对应的值是该A-mer在所有测序片段中出现的次数和含有该hmer的测序片段的ID。为了减少内存的使用,我们实际上并不真正存储A:mer的序列,而是将kmer存储为一个64位的无符号整数,其中每两个位编码一个核苷酸,因此hmer的长度最大值为32。
作为种子的it-mer需要满足以下三个要求:
(a)该hmer的信息摘#>1.5(在本章最后一节中会详细介绍信息熵的概念以及如何计算);(b)该tmer在测序数据中至少出现两次;(c)该hmer不是回文结构。所谓回文机构,是指结构相同、方向相反的序列。例如,以下两个序列:
我们采取一种贪婪的策略从一个作为种子的hmer出发,逐步构建一个剪接图,具体步骤如下:
(1)在当前哈希表中选择一个出现频率最大的并且可以作为种子的hmer,当作一个剪接图最初的主干序列。
(2)在所有未使用的A:-mer中找出一个其k-l前缀跟当前主干序列的5’端的免-mer的kA后缀完全相同的、并且频率最高的A-mer,利用此A:-tner将当前主干序列向5’端延长一个核苷酸,之后继续按照这种方式延长到不能够延长为止,类似地对向主干序列3’端方向进行延长。
(3)利用双端测序的信息(如果可用的话),进一步延长当前剪接图的主干序列。利用哈希表可以很容易的获得映射到主干序列端点的测序片段,检查它们对应的另一端的测序片段是否在当前的图的拼接过程中被己经被使用到,如果没有,说明该序列并不完整。这时我们通过另一端的测序序列延长出另一半序列,跟当前主干连接起来(图3.3c)。
(4)对于当前剪接图的每一个A:-mer,检查是否存在某一个A-mer可以被其它未用到的A:-mer继续延长(例如图3.7a中左侧的红色的hmerATCAG)。我们将这样的A:-mer称为分支A:-nier。一旦发现一个分支A:-mer,从这个yt-mer出发,按照以上的方式继续去延长出一个新的序列。如果新延长出来的序列能重新贴回到主干序列上,那么就发现了一个可变剪接,这时更新当前的剪接图(图3.7b)。否则,新延长出来的序列就是一个图中一个潜在的分支。这时需用一些准则来检验这个潜在的分支是由于可变剪接造成的真正的分支还是由于测序错误
造成的假的分支。重复此步骤,直到没有分支hmer为止。
(5) 一个剪接图构造完成后,记录下图中所有用到的所有hmer,将来构造别的剪接图的过程中尽量不再去使用这些yt-mer。
(6)把测序片段映射到剪接图中,来修剪图中的边,去掉那些因为测序错误导致的边。这一点通过我们的哈希表很容易实现。
(7)在所有未使用的it-mer中选择一个频率最高的作为新的种子,重复以上步骤,直到整个哈希表中的tmer全部被使用。
3.3.2构造兼容图
我们仔细分析了剪接图的结构发现,转录体的拼接可以仅仅依靠剪接图中的边来完成,其原因并不能严格进行数学证明,但是我们可以利用图3.7给的一个简单例子得到一个直观的感受。从图3.7的例子中可以看到,该基因的两个转录体,其中的一个可以依靠图中黄色的外显子唯一标识出,但是另一个转录体却没法依靠任何外显子去唯一标识,也就是说,仅仅依靠外显子无法唯一标识出基因的每一个转录体。
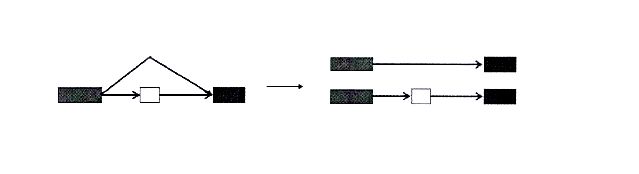
然而,换一个角度去看,不去关注外显子,而是关注外显子和外显子的组合,问题就迎刃而解了。两个转录体所含有的外显子的组合的不同恰恰唯一标识了不同的转录体,例如图3.7中的例子,红色边代表的外显子组合和蓝色边代表的外显子组合分别对应了两个不同的转录体。也就是说,对于发生可变剪接的基因,其它的一个转录体可能并没有不同于其他转录体的外显子,但是往往有一个不同于转录体的外显子组合图3.7.转录体的拼接可以仅仅依靠剪接图中的边的组合来实现。图中的基因对应着两个转录体,可以靠图中的两种颜色的边分别标识出来。
从上面的讨论可以看出,转录组的拼接问题可以转化为剪接图中边的组合问题,如果能给出剪接图中边的一个好的组合,就能给出正确的转录体。剪接图中的每一条边都应该至少存在于一个转录体中。我们希望能找到一个转录体的集合,该集合中的转录体序列能够覆盖剪接图中的每一条边,并且使该集合中的转录体数目尽可能的少。
不难发现,剪接图中的路跟兼容图中的路是一一对应的,也就是说每一个转录体不仅对应着剪接图中的一条路,也对应着兼容图中的一条路。这样,我们把转录组拼接问题又转化成了在兼容图中找一组路,使得能覆盖兼容图的每一点,而且路的数目尽可能少。很自然地,我们想到了一个经典的组合优化模型一一最小路覆盖模型。
但是并不是剪接图或者兼容图中的每一条路都对应一个真正的转录体。因此,必须寻找一种方式,确保来自同一个转录体的连接被组合到一起。为此,我们给兼容图加权:对于兼容图中的每一个点X,其实对应着剪接图中的一条弧(e’,e"),我们给X定义了两个权重:“出”权重W、o和“入”权重IV"。出权重跨越弧(e’,e’’)的测序片段的数目和跨越所有以e’为弧尾的弧(包括(e',e")本身)的测序片段的总数目的比值。入权重Fx,,?是跨越弧(e’,e’’)的测序片段的数目和跨越所有以e’’为弧头的弧(包括(e’,e’’)本身)的测序片段的总数目的比值。
3.3.3寻找最优的转录体集合
为了应用最小路覆盖模型,我们首先计算兼容图C的传递闭包G。然后,定义一个二部图,叫做可达性(reachability)图。图3.9给出了一个传递闭包和对应的可达性图的例子。
图3.9.由兼容图构造的传递闭包以及由传递闭包定义的可达性图的示意图。对于图G中的每一个顶点X,我们在二部图中定义两个点Ix,凡.,这两个点分别在左右两个分部中;二部图中的从到之间连一条边当且仅当在G中存在一条(x,_y)边。
边上的权重定义如下:
W^=-\o^\-\W,,-W^,^I)
其中Wx,i和Wy’o分别是图C中点;C的入权重和>;的出权重。定义了权重之后,在这个二部图中计算最小权重的最大匹配。根据Dilworth定理,可以从这个匹配中得到兼容图的最小路覆盖,进一步扩展为一个路的集合。注意到兼容图和剪接图的对应关系,兼容图中路可以很容易转化为剪接图中的路,从而给出转录体的集合。
第四章Bridger的测评
4.1测试数据和评价标准
将Bridger跟其它的从头拼接算法进行比较,这里选取的其它从头拼接算法包括ABySS(.version1.3.4),Trinity (version2012-10-25) , Oases{version 0.2.02),其中Oases需要用到一个基因组拼接工具Velvet (version1.2.01)]。Oases和Bridger中,hmer的长度不仅可以取一个单一的值,还可以取多个值然后对不同A值的拼接结果进行合并,为了区分,将它们利用多个;t值的拼接结果分别记为Oases_M和Bridger一M。算法Trans-ABySS 也是采用多个yt的策略,但是很多研究表明它的表现不如Oases—MM,因此这里的比较没有将Trans-ABySS包含进来。另外,为了更好地验证Bridger的结果,我们将从头拼接算法Cufflinks [68]也拿来一起比较,当作评价从头拼接算法的一个很好的标尺。本节对测试中用到的三个RNA测序数据、不同算法在不同数据测试时的参数设置以及评价不同算法优劣的标准进行了详细的描述。
4.1.1测试的数据
通过对老鼠,狗,人的测试数据去探讨这一问题,在测试中,我们所用的数据都可以通过NCBI或者DDBJ的SRA数据库下载到。狗的RNA 测序数据来自于最近的一项研究(Liu et al. Spontaneously occurring canine mammary cancers faithfully represent their human counterparts,尚未发表)’该数据大约有三千万个长度为50bp的双端测序序列片段,中间的插入长度为130bp(Accession Code: SRR882093)。人的RNA测序数据来自于人的CD4 T细胞,有五千万个长度为45bp的双端测序序列片段,中间的插入长度为200bp(Accession Code: SRX011545, SRX011546)。老鼠的测序数据是 Trinity 所用的一个测试数据[42],该数据具有链特异性,大约五千万个长度是76bp的双端测序序列片段,中间插入长度是300bp (Accession Code: SRX062280)
4.1.2参数设置
不同的拼接算法提供的参数不同,在测试中,为了让每个拼接算法都能达到最好的拼接效果,需要将各个算法的参数进行优化,优化后各个算法的设置如下
(对于这里没有特别指明的参数均采用默认参数):
(1) ABySS;“ abyss-pe k=25 c=2 E=0 j=6 in=‘left.fq right.fq ”’。对于狗和人的数据,A:-mer的长度取25,对于老鼠的数据,的值取33以便能够得到更好的效果。
(2) Trinity:“seqTypefq--CPU 6—bflyHeapSpaceMax lOG —hflyGCThreads4”。对于老鼠的数据,需要加上参数“--SS—lib type RF”。Trinity中A:的值只能是固定值25。
(3) Oases: “-ins—kngth 200 -cov—cutoff 2 -edgeFractionCutoff 0.05,对于狗和人的数据,;fc-mer的长度取25,对于老鼠的数据,取31能够得到
(4) Bridger: ''—seqTypefq —kmerjength 25 —CPU6'\ 同 Oases—样,在狗和人的数据上A=25,在老鼠的数据上A:=31。另外,对于老鼠的数据,也需要加上参数“--SSjibjypeRF’’,更好的利用链的特异性信息。
(5) Oases一M:在狗和人的数据上,的不同取值为21,25,33, 37,43;在老鼠的数据上,A取25,31, 37, 43, 49。
(6) Bridger_M:由于Bridger中A:不得超过32,因此在狗和人的数据上,k取值为21, 23,25,27,29;而在老鼠的数据上,的取值为23,25,27,29, 31。
(7) Cufflinks:默认参数。
4.2评价方法和标准
对真实的转录组测序数据的拼接结果进行评价是一个不太容易的问题,主要是因为我们并不知道当前转录组中表达的基因是哪些,更不知道表达的转录体是什么。基因组拼接的一些评价标准,对于转录组并不太适用。
虽然对于转录体拼接的评价,目前还没有统一的标准,但是我们还是可以通过利用己知的转录体对转录组的拼接在一定程度上进行合理的评价。我们将当前己知的转录体称为参考转录体,将一个拼接算法拼接出来的转录体称为候选转录体。直观地,如果一个候选转录体跟一个参考转录体一模一样,则我们基本可以断定这个拼接出来的转录体是正确的。
当然,在实际中要求两个序列完全一模一样过于苟刻,通常允许存在一定的误差存在。当前己知的转录体数据是可以拼接出来的所有转录体利用BLAT跟所有的参考转录体进行联配,序列的相似性参数设置为95%。如果一个拼接出来的转录体能完全覆盖某个参考转录体,并且插入和删除的碱基不超过序列长度的1%,就称这个参考转录体被全长(ftill-length)重构了出来,并把这个拼接出来的转录体叫做一个真阳性的转录体。类似地,可以定义被80%重构出来的参考转录体,即该参考转录体的序列的80%被某个拼接出来的转录体覆盖。从不同的水平上进行评价,可以对不同的拼接算法进行更好的比较。值得说明的一点是,这里对插入和删除的要求较严格,主要是考虑到复杂的真核生物中可变剪接的原因,如果不严格限制插入和删除,会很容易造成错误的评价。
(2) Bridger使用了一个严格的数学模型在图中搜索一个最优的路的集合,跟Trinity的穷举法相比,大大降低了拼接结果中的假阳性。更重要的是,假阳性的降低并没有以牺牲敏感性为代价。
(3) Bridger是第一个成功地将测序深度信息整合到从头拼接中的算法,这一信息大大帮助了我们在图中更准确地找对应转录体的路,是Bridger超过其它算法的一个重要原因。
(4) Bridger没有去建立一个deBruijn图,而是直接由测序片段构建剪接图,并且在构建图的过程中利用了双端测序的信息,一方面来帮助我们建立一个更完整的图,为完整的拼接提供保障;另一方面帮助我们有效地控制了图的大小,极大降低了以后在图中找路的难度,提高了计算的效率。
(5) Bridger的算法在理论上是一个多项式的算法,而Trinity的算法是一个指数时间算法。在实际中,Bridger的运行时间和需要的内存都比 Trinity 少。尽管Bridger有很多优点,但是对比实际运行时间和CPU时间不难看出,它的效率还有待提高。例如,Bridger的CUP时间大约是Trinity的1/4,但是实际运行时间却没有这么大的优势。在狗和人的数据上,Bridger的运行时间大约是Trinity的一半,在老鼠的数据上,Bridger的运行时间大约是Trinity的3/4。
因此,Bridger的代码实现还值得进一步优化。尽管优化代码并不涉及算法方面,但还是很重要的。Trinity的现在版本跟最初版本相比,通过对程序进行高度的并行化,运行时间己经提高了十来倍。Bridger的CPU时间跟ABySS是差不多的,所以,Bridger经过优化后的运行时间至少可以达到跟ABySS相当,如果那样,Bridger就更加完美了,几乎在任何方面都超过了其它从头拼接算法。Bridger的高度并行化是我们将来一个努力的方向。Bridger的最小路覆盖模型尽管很好,但也不是万能的。有时候,由于它一味地追求用最小数目的路来解释当前看到的剪接图,可能会导致漏掉某些转录
4.3 Bridger的优缺点
Bridger给转录组的从头拼接提供了一个更好的解决办法,对将来转录组的研究有重要的意义。跟当前最好的从头拼接算法Trinity相比,Bridger主要有以下几个优点:
(1) Trinity使用了一个固定的A:值(A: = 25),虽然大多数情况是表现还不错,但是一个固定的不可能对每一个数据来说都是最好的选择;而在Bridger中,用户可以根据不同的数据特点来变化的值,从而得到更好的效果。
例如,如果某个转录体的序列是另一个转录体序列的一个子序列,那么根据最小路覆盖模型,只能给出较长的转录体,而漏掉了短的转录体。当然,我们可以通过一些技巧来弥补这一缺陷。我们可以对拼接处的转录体的不同位置进行测序深度的检查,如果某个转录体前后不同位置的测序深度相差很大,极可能还存在一个短的转录体,其序列是这个转录体的一个子序列。在敏感性方面,Bridger比当前所有的从头拼接算法都要好,在三个测序数据上都全长重构出了更多的参考转录体。
有趣的是,当前最好的从头拼接算法Trinity中利用了穷举法在图中寻找对应转录体的路,理论上敏感性己经达到一个比较好的程度,但是三个测试数据的结果都表明Bridger的敏感性比Trinity还要好。虽然在人的数据上Bridger敏感性还不如Cufflinks好,但是在狗的数据上Bridger的敏感性跟Cufflinks相差不大,在老鼠的测试数据上Bridger的敏感性甚至比Cufflinks还要好。值得一提的是,BridgerJVI的敏感性更高,在狗和老鼠的数据上都超过了 Cufflinks。其它的拼接算法,如ABySS、Oases和Oases_M,即使跟Trinity相比,敏感性也相差很多。另外,我们以80%被重构出来的参考转录体为标准进行比较,同样可以观察到相似的结果,并且Bridger的优势更加明显。
值得一提的是,Bridget预测的候选转录体的数目比其它从头拼接算法少的多,通常少预测了 10000到30000个候选转录体,但是却拼接出了更多的参考转录体,具有更高的准确性,说明Bridger的假阳性跟其它方法相比,特别是跟Trinity相比,假阳性大大降低了。这正是Bridger最突出的贡献。
对于转录组拼接,计算资源的需求往往很大,因此也是很重要的一个方面评价标准。在拼接过程中,我们分别记录了不同的拼接工具的运行时间、CPU时间和内存使用情况。这里,我们只考虑四个使用单个值的从头拼接算法,不再考虑多个k的拼接算法Oases_M和Bridger_M,也不考虑基于参考基因组的算法Cufflinks,这些拼接算法通常分为多个步骤去完成,中间有间断,不适合拿来比较。为了比较的公平性,我们将A:值统一设定为25,因为Trinity中&值只能取25。
ABySS在内存和运行时间方面表现最好;Oases的表现很不稳定,在狗的数据上使用的时间和内存较少,但是在人和老鼠的数据上使用了最大的内存,并且运行时间也很长;Trinity的表现比较稳定,但是运行时间很长;Bridget"不仅在内存和运行时间上都比较稳定,并且在运行时间和使用的内存上比Oases和Trinity都要好。
总体来讲,Bridger在敏感性和准确性方面都超过了当前所有其它的从头拼接算法,是从头拼接有了很大的进步和提高。虽然在运行时间和内存使用上比ABySS稍差,但是远远好于其它的从头拼接算法。另外,ABySS是一个基因组拼接算法,在转录组拼接中效果很差,考虑到这个因素,Bridger可以说是转录组拼接时是最好的选择。
4.4 Bridger的下游分析
拼接出一个转录组之后,可以发现很多新的基因和转录体,可以发现某一个基因新的可变剪接方式,但是转录组研究中生物学家或者医学家更为关心的是,如何进一步确定出这些转录体的表达丰度(abundance),如何确定不同样本中表达有差异的基因和转录体,这些研究往往更有实际的意义,包括癌症在内的许多疾病都与转录体的差异表达有关。本节将介绍如何利用Bridger的拼接结果去做这些进一步的研究。
利用RNA测序数据数据来估计表达丰度的研究很多,最初对于基因的表达丰度的估计仅仅是通过统计映射到每个基因上的测序片段的个数来实现的,但是这种简单的方法对估计转录体的表达并不好,因为可变剪接的存在,根本无法准确区分一个测序片段来自于基因的哪一条转录体。
因此,人们开始通过更复杂的统计方法来对转录体的表达丰度的估计进行建模,并开发了很多方法和工具。但是,大部分方法都是依赖参考基因组或者基因组的一些注释信息,并不太适合从头拼接。最近,Li 等人设计了一个利用 EM (Expectation Maximization)算法来估计转录体表达丰度的工具RSEM,不需要借助基因组信息,因此特别适合从头拼接的转录体的表达丰度估计。RSEM可以对每个基因的每个转录体都出一个转达水平的估计。估计出不同的转录体的表达值后,还可以根据表达值来过滤掉错误拼接的一些转录体。
例如,一个基因有多个转录体,如果其中的一个转录体的表达极低,就很有可能是一个错误的拼接,当然如何去确定一个阈值来进行过滤也是需要研究的问题。我们的建议是不要直接删除掉过滤出来的转录体,而是放到一边,以备将来的研究中可能用得到。
第五章 总结
在本文中,我们主要是针对基因转录组的从头拼接问题,去探究并设计了一个具有高效性特点的算法——Bridger算法,在Bridger算法中,我们通过对一些生物的RNA测序数据上的测试结果,探析Bridger算法的优缺点,不过从优缺点看,算法Bridger比目前使用的一些算法都还要成功,拼接效果也要优于所有的从头拼接算法,Bridger算法不仅可以能拼接出相对精确的转录体,它还能给的候选转录体的数目要比其它拼接方法给的候选转录体少,因此,利用该方法拼接,可以极大限度地降低了拼接结果中的假阳性。
更为有趣的是,在大多数情况下,Bridger算法甚至可以跟基于参考基因组的拼接工具Cufflinks相媳美。而在计算资源方面,理论上,算法Bridger的CPU运算时间仅仅相当于当前最好的从头拼接算法Trinity,这是理论上,而实际上,此算法的运行时间要比算法Trinity快的多,除运算的时间短之外,在内存上,该算法所占的内存相对于其他算法的内存还要少。
在文中,我们通过一些实际的例子,展现出Bridger算法在转录组研究中的重要地位与适用价值以及对Bridger算法拼接结果的下游,作一定的研究分析,在表达丰度的估计与差异性表达的研究分析时,在目前,我们已经拥有诸多的算法与研究工具,研究者们也可以根据不同的研究目的去挑选择一些较为合适的研究工具,从而达到对转录组的深入研究分析。
第一代测序技术与第二代测序技术在科技的冲击下,逐步取得良好的发展,最终,第三代测序技术己在此基础上横空出世。在新产生的第三代测序技术中,其最大特点是测序片段较长,甚至可以达到几千个甚至几万个碱基,测序片段的长度长对于转录体而言,冲击效果十分明显,长度较长的测序片段在基因中基本上可以覆盖整个转录体。但是,目前该技术还面临很大的技术瓶颈,主要是测序错误率太高,而且通量比较低,因此应用价值还十分有限。我们相信,在一段相当长的时间内,本文提出的Bridger算法将会对转录组的研究起到巨大的推动作用。
在本文中,新算法Bridger几个特点:第一,放弃了通常使用的deBruijn图,由RNA的测序片段来直接构建一个能更好地反映出每一个基因可变剪接结构的图一一剪接图。
第二,构造图的过程中利用双端测序的信息,不仅使得到的剪接图更加准确、完整,而且有效地控制了图的规模,从而降低了在图中寻找对应转录体的路的难度。第三,通过引进一个辅助图,成功地将一个经典的组合优化模型应用到转录组的从头拼接中,相比于以前的穷举方法,可以大大降低结果的假阳性。通过给模型加权,巧妙地将测序的深度信息整合到模型中,大大提高了拼接的准确性,据我们所知,这是测序的深度信息第一次被成功地用在从头拼接算法中。尽管Bridger算法有很多优点,但是也存在不足。
第一,当前的Bridger的代码实现还有待进一步优化,在构造剪接图的过程中实现并行化计算是我们的一个努力方向。
第二,算法中的最小路覆盖模型,并不是对于任何情况都非常有效,有些比较特殊例子,该模型也表现不太理想,这时可以通过一些技巧来克服算法的不足。
对于这些不足之处,还需我们在今后的学习研究中逐步探寻,继而一一化解。
参考文献:
[1] Lockhart DJ, Winzeler EA. Genomics, gene expressionand DNA arrays. Nature, 2000, 405(6788): 827–836.
[2] Costa V, Angelini C, De Feis I, Ciccodicola A. Uncovering the complexity of transcriptomes with RNA-Seq. J BiomedBiotechnol, 2010, 2010: 853916.
[3] Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionarytool for transcriptomics. Nat Rev Genet, 2009, 10(1): 57–63.
[4]许波,张伟强,冯晓曦,等.转录组测序技术在玉米中的应用研究进展[J].玉米科学,2014,22(1):67~72,78.
[5] Maher CA,Kumar- sinha C,Cao XH,etal.Trancriptome Sequencing to detect gene fusions in cancer.Nature,2009,458 (7234):97一101.
[6] 周晓光,任鲁风,李运涛,等.下一代测序技术:技术回顾与展望[J].中国科学生命科学,2010,40(1);23-37
[7] . Zhou X G, Ren L F, Li Y T, et al. The next-generation sequencingtechnology: a technology review and future perspective[J].ScientiaSinicaVitae, 2010,40(1): 23-37.
[8] Schuster S C.Next-generation sequencing transforms today's biology [J]. Nature, 2008, 200(8): 16-18.
[9] Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool fortranscriptomics[J]. Nature Reviews Genetics, 2009, 10(1): 57-63.
[10] 杨晓玲,施苏华,唐恬.新一代测序技术的发展及应用前景[J].生物技术通报,2010(10):76-81 .
[11]张春兰,秦孜娟,王桂芝,等.转录组与RNA一Seq技术.生物孩术通报,2012年第12期,
[12]祁云霞,刘永斌,荣威恒.转录组研究新技术:RNA一seq及其应用.遗传,2011,33(11):1191一1202.
[13] Ali Mottazavi, Williams BA, McCue K, et al. Mapping and quantifying mammaliantranscriptomesbyRNA-seq. Nat Methods, 2008,5(7):621-8.
[14] Maher CA, Sinhal CK, Cao XH, et al. Transcriptomesequencing to detect gene fusions in cancer. Nature, 2009, 458: 97-101.
[15] Sergei A. Filichkin, Henry D. Priest, Scott A. Givan, etal. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. GenomeRes, 2010,20:45-58. Vliet VA. Next generation sequencing of microbial transcriptomes: challenges
[16]李小白, 张明龙, 崔海瑞. 油菜EST资源的SSR信息分析. 中国油料作物学报(Li Xiaobai, Zhang Minglong, Cui Hairui. Analysis of SSR information in EST resource of oilseed rape Chinese. Journal of Oil Crop Sciences) 2007; 29(1): 20-5.
[17]Yu JK, La Rota M, Kantety RV, Sorrells ME. EST derived SSR markers for comparative mapping in wheat and rice. Mol Genet Genomics 2004; 271 (6): 742-51.
[18]吴磊,王丹,苏文悦,郭长虹,束永俊.利用比较基因组学开发山羊草属Idle分子标记.作物学报.
(Wu Lei, Wang Dan, Su Wenyue, Guo Changhong, Shu Yongjun. Developing InDel markers from aegilops genus based on comparative genomics. Acta Agronomica Sinica) 2012; 38 (7): 1334-8.and opportunities. FEMS Microbiol Lett, 2010, 302(1): 1–7.
[17] Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionarytool for transcriptomics. Nat Rev Genet, 2009, 10(1):57–63. [4] 454 Home Page.
[18] Tang FC, Barbacioru C, Wang YZ, Nordman E, Lee C, XuNL, Wang XH, Bodeau J, Tuch BB, Siddiqui A, Lao KQ, Surani MA. mRNA-Seq
whole-transcriptome analysis of asingle cell. Nat Methods, 2009, 6(5): 377–382.
[19]杨晓玲,施苏华,唐恬.新一代测序技术的发展及应用前景[J].生物技术通报,2010(10):76-81 .
[20] Croucher NJ, Fookes MC, Perkins TT, Turner DJ, MargueratSB, Keane T, Quail MA, He M, Assefa S, Bähler J, Kingsley RA, Parkhill J, Bentley SD, Dougan G, ThomsonNR. A simple method for directional transcriptome sequencing using Illumina technology. Nucleic Acids Res, 2009, 37(22): e148.
[21] Vivancos AP, Güell M, Dohm JC, Serrano L, HimmelbauerH. Strand-specific deep sequencing of the transcriptome.Genome Res, 2010, 20(7): 989–999.
[22] 李滢, 孙超, 罗红梅, 李西文, 牛云云, 陈士林. 基于高通量测序454 GS FLX的丹参转录组学研究. 药学学报, 2010, 45(4): 524–529.
[23]朱国念,等.克百威残留检测直接竞争ELISA试剂盒的研究.中国食品学报,2003,3(3):1-6.
[24]张英华,迟玉杰.酶联免疫法测定牛初乳中乳铁蛋白含量.中国乳品工业,1999,27(6):19-21.
[25]张爱霞,张佳程,生庆海.牛乳中纤维素蛋白酶及其酶原活性测定的比较.中国乳业,2003(2):39-40.
[26]张国龙,李德发.大豆蛋白酶抑制因子酶联免疫吸附测定方法的研究.动物营养学报,1997,9(1):12-20.
[27]AnklamE,GadaniF,HeinzeP,eta.lAnalyticalmethodsforde-tectionanddeterminationofgeneticallymodifiedorganismsinagr-iculturalcropsangplants-derivedfoodproducts.EurFoodResTechno,l2002,214:3-26.
[28] Der JP, Barker MS, Wickett NJ, Depamphilis CW, Wolf PG. De novo characterization of the gametophyte transcriptome in bracken fern, Pteridium aquilinum. BMC Genomics, 2011, 12(1): 99.
[29] Logacheva MD, Kasianov AS, Vinogradov DV, Samigullin TH, Gelfand MS, Makeev VJ, Penin AA. De novo sequencing and characterization of floral transcriptome in two species of buckwheat (Fagopyrum). BMC Genomics, 2011, 12(1): 30.
[30] Rokyta DR, Wray KP, Lemmon AR, Lemmon EM, Caudle SB. A high-throughput venom-gland transcriptome for the Eastern Diamondback Rattlesnake (Crotalus adamanteus) and evidence for pervasive positive selection across toxin classes. Toxicon, 2011, 57(5): 657-671.
[31] Johansen SD, Karlsen BO, Furmanek T, Andreassen M, Jørgensen TE, Bizuayehu TT, Breines R, Emblem Å, Kettunen P, Luukko K. RNA deep sequencing of the Atlantic cod transcriptome. Comp Biochem Physiol Part D Genomics Proteomics, 2011, 6(1): 18-22.
[32] Swarbreck SM, Lindquist EA, Ackerly DD, Andersen GL. Analysis of leaf and root transcriptomes of soil-grown Avena barbata plants. Plant Cell Physiologic, 2011, 52(2): 317-332
[33] Gregory R, Darby AC, Irving H, Coulibaly MB, Hughes M, Koekemoer LL, Coetzee M, Ranson H, Hemingway J, Hall N. A de novo expression profiling of Anopheles funestus, malaria vector in Africa, using 454 pyrosequencing. PLoS ONE, 2011, 6(2): e17418.
[34] Blanca J, Canizares J, Roig C, Ziarsolo P, Nuez F, Pico B. Transcriptome characterization and high throughput SSRs and SNPs discovery in Cucurbita pepo (Cucurbitaceae). BMC Genomics, 2011, 12(1): 104.
[35] Garg R, Patel RK, Tyagi AK, Jain M. De novo assembly of chickpea transcriptome using short reads for gene discovery and marker identification. DNA Res, 2011, 18(1): 53-63.
[36] Sun X, Zhou S, Meng F, Liu S. De novo assembly and characterization of the garlic (Allium sativum) bud transcriptome by Illumina sequencing. Plant Cell Rep, 2012, 31(10): 1823-1828.
[37] Gao X, Han J, Lu Z, Li Y, He C. Characterization of the spotted seal( Phoca largha) transcriptome using Illumina paired-end sequencing and development of SSR markers. Comp Biochem Physiol Part D Genomics Proteomics, 2012, 7(1): 277-284.
[38] Peterson MP, Whittaker DJ, Ambreth S, Sureshchandra S, Buechlein A, Podicheti R, Choi JH, Lai Z, Mockatis K, Colbourne J. De novo transcriptome sequencing in a songbird, the dark-eyed junco (Junco hyemalis): genomic tools for an ecological model system. BMC Genomics, 2012, 13(1): 305.
[39] He M, Wang Y, Hua W, Zhang Y, Wang Z. De novo sequencing of Hypericum perforatum transcriptome to identify potential genes involved in the biosynthesis of active metabolites. PLoS ONE, 2012, 7(7): e42081.
[40] Wang B, Ekblom R, Castoe TA, Jones EP, Kozma R, Bongcam-Rudloff E, Pollock DD, Hoglund J. Transcriptome sequencing of black grouse (Tetrao tetrix) for immune gene discovery and microsatellite development. Open Biol, 2012, 2(4): 120054
[41] Lulin H, Xiao Y, Pei S, Wen T, Shangqin H. The first Illumina-based de novo transcriptome sequencing and analysis of safflower flowers. PLoS ONE, 2012, 7(6): e38653.
[42] Gahlan P, Singh HR, Shankar R, Sharma N, Kumari A, Chawla V, Ahuja PS, Kumar S. De novo sequencing and characterization of Picrorhiza kurrooa transcriptome at two temperatures showed major transcriptome adjustments. BMC Genomics, 2012, 13(1): 126.
[43] Nyberg KG, Conte MA, Kostyun JL, Forde A, Bely AE. Transcriptome characterization via 454 pyrosequencing of the annelid Pristina leidyi, an emerging model for studying the evolution of regeneration. BMC Genomics, 2012, 13(1): 287.
[44] Zhou Y, Gao F, Liu R, Feng J, Li H. De novo sequencing and analysis of root transcriptome using 454
pyrosequencing to discover putative genes associated with drought tolerance in Ammopiptanthus mongolicus. BMC Genomics, 2012, 13(1): 266.
[45] Yang F, Xu B, Zhao S, Li J, Yang Y, Tang X, Wang F, Peng M, Huang Z. De novo sequencing and analysis of the termite mushroom (Termitomyces albuminosus) transcriptome to discover putative genes involved in bioactive component biosynthesis. J Biosci Bioeng, 2012, 114(2): 228-231.
[84] Van Belleghem SM, Roelofs D, Van Houdt J, Hendrickx F. De novo Transcriptome assembly and SNP discovery in the wing polymorphic salt marsh beetle Pogonus chalceus (Coleoptera, Carabidae). PLoS ONE, 2012, 7(8): e42605.
[46] Torales SL, Rivarola M, Pomponio MF, Fernandez P, Acuna CV, Marchelli P, Gonzalez S, Azpilicueta MM, Hopp HE, Gallo LA, Paniego NB, Marcucci Poltri SN. Transcriptome survey of Patagonian southern beech Nothofagus nervosa (= N. alpina): assembly, annotation and molecular marker discovery. BMC Genomics, 2012, 13(1): 291.
[47] Sadamoto H, Takahashi H, Okada T, Kenmoku H, Toyota M, Asakawa Y. De novo sequencing and transcriptome analysis of the central nervous system of mollusc Lymnaea stagnalis by deep RNA sequencing. PLoS ONE, 2012, 7(8): e42546.
[48] Hsu JC, Chien TY, Hu CC, Chen MJ, Wu WJ, Feng HT, Haymer DS, Chen CY. Discovery of genes related to insecticide resistance in Bactrocera dorsalis by functional genomic analysis of a de novo assembled transcriptome. PLoS ONE, 2012, 7(8): e40950.
[49] Tanase K, Nishitani C, Hirakawa H, Isobe S, Tabata S, Ohmiya A, Onozaki T. Transcriptome analysis of carnation (Dianthus caryophyllus L.) based on next-generation sequencing technology. BMC Genomics, 2012, 13(1): 292.
[50] Ji P, Liu G, Xu J, Wang X, Li J, Zhao Z, Zhang X, Zhang Y, Xu P, Sun X. Characterization of common carp transcriptome: Sequencing, de novo assembly, annotation and comparative genomics. PLoS ONE, 2012, 7(4): e35152.
[51] Werner GD, Gemmell P, Grosser S, Hamer R, Shimeld SM. Analysis of a deep transcriptome from the mantle tissue of Patella vulgata Linnaeus (Mollusca: Gastropoda: Patellidae) reveals candidate biomineralising genes. Mar Biotechnol (NY), 2013: 15(2):230-243.
[52] Marra NJ, Eo SH, Hale MC, Waser PM, Dewoody JA. A priori and a posteriori approaches for finding genes of evolutionary interest in non-model species: Osmoregulatory genes in the kidney transcriptome of the desert rodent Dipodomys spectabilis (banner-tailed kangaroo rat). Comp Biochem Physiol Part D Genomics Proteomics, 2012, 7(4): 328-339.
[53] Ma K, Qiu G, Feng J, Li J. Transcriptome analysis of the oriental river prawn, Macrobrachium nipponense using 454 pyrosequencing for discovery of genes and markers. PLoS ONE, 2012, 7(6): e39727. [93] Huh JW, Kim YH, Park SJ, Kim DS, Lee SR, Kim KM, Jeong KJ, Kim JS, Song BS, Sim BW, Kim SU, Kim SH, Chang KT. Large-scale transcriptome sequencing and gene analyses in the crab-eating macaque (Macaca fascicularis) for biomedical research. BMC Genomics, 2012, 13(1): 163.
[54] Hao D, Ma P, Mu J, Chen S, Xiao P, Peng Y, Huo L, Xu L, Sun C. De novo characterization of the root transcriptome of a traditional Chinese medicinal plant Polygonum cuspidatum. Sci China Life Sci, 2012, 55(5): 466.
[55] Blanca J, Esteras C, Ziarsolo P, Perez D, Fernandez V, Collado C, Rodriguez R, Ballester A, Roig C, Canizares J, Pico B. Transcriptome sequencing for SNP discovery across Cucumis melo. BMC Genomics, 2012, 13(1): 280[56] Kaur S, Pembleton LW, Cogan NO, Savin KW, Leonforte T, Paull J, Materne M, Forster JW. Transcriptome sequencing of field pea and faba bean for discovery and validation of SSR genetic markers. BMC Genomics, 2012, 13(1): 104.
[57] Gao Z, Luo W, Liu H, Zeng C, Liu X, Yi S, Wang W. Transcriptome analysis and SSR/SNP markers information of the blunt snout bream (Megalobrama amblycephala). PLoS ONE, 2012, 7(8): e42637.
[58] Xie F, Burklew CE, Yang Y, Liu M, Xiao P, Zhang B, Qiu D. De novo sequencing and a comprehensive analysis of purple sweet potato (Impomoea batatas L.) transcriptome. Planta, 2012, 236(1): 101-113.
[59] Pereiro P, Balseiro P, Romero A, Dios S, Forn-Cuni G, Fuste B, Planas JV, Beltran S, Novoa B, Figueras A. High-throughput sequence analysis of turbot (Scophthalmus maximus) transcriptome using 454-pyrosequencing for the discovery of antiviral immune genes. PLoS ONE, 2012, 7(5): e35369. [100] Li Y, Sun C, Luo H, Li X, Niu Y, Chen S. Transcriptome characterization for Salvia miltiorrhiza using 454 GS FLX]. Acta Pharmaceutica Sin, 2010, 45(4): 524-529.
[60] Kumar S, Blaxter ML. Comparing de novo assemblers for 454 transcriptome data. BMC Genomics, 2010, 11(1): 571.
[61] Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, Berka J, Braverman MS, Chen YJ, Chen Z, Dewell SB, Du L, Fierro JM, Gomes XV, Godwin BC, He W, Helgesen S, Ho CH, Irzyk GP, Jando SC, Alenquer ML, Jarvie TP, Jirage KB, Kim JB, Knight JR, Lanza JR, Leamon JH, Lefkowitz SM, Lei M, Li J, Lohman KL, Lu H, Makhijani VB, McDade KE, McKenna MP, Myers EW, Nickerson E, Nobile JR, Plant R, Puc BP, Ronan MT, Roth GT, Sarkis GJ, Simons JF, Simpson JW, Srinivasan M, Tartaro KR, Tomasz A, Vogt KA, Volkmer GA, Wang SH, Wang Y, Weiner MP, Yu P, Begley RF, Rothberg JM. Genome sequencing in microfabricated high-density picolitre reactors. Nature, 2005, 437(7057): 376-380.
[61] Chevreux B, Wetter T, Suhai S. Genome sequence assembly using trace signals and additional sequence information. in Computer Science and Biology: Proceedings of the German Conference on Bioinformatics (GCB). 1999.
[62] Huang X, Madan A. CAP3: A DNA sequence assembly program. Genome Res, 1999, 9(9): 868-877. [105] Swindell SR, Plasterer TN. SEQMAN. Contig assembly. Methods Mol Biol, 1997, 70: 75-89.
[106] Pertea G, Huang XQ, Liang F, Antonescu V, Sultana R, Karamycheva S, Lee Y, White J, Cheung F, Parvizi B, Tsai J, Quackenbush J. TIGR Gene Indices clustering tools (TGICL): a software system for fast clustering of large EST datasets. Bioinformatics, 2003, 19(5): 651-652.
[63] Miller RT, Christoffels AG, Gopalakrishnan C, Burke J, Ptitsyn AA, Broveak TR, Hide WA. A comprehensive approach to clustering of expressed human gene sequence: the sequence tag alignment and consensus knowledge base. Genome Res, 1999, 9(11): 1143-1155.
[64] Zerbino DR, Birney E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res, 2008, 18(5): 821-829.
[65] Jackman SD, Birol I. Assembling genomes using short-read sequencing technology. Genome Bicol, 2010, 11(1): 202.
[66] MacCallum I, Przybylski D, Gnerre S, Burton J, Shlyakhter I, Gnirke A, Malek J, McKernan K, Ranade S, Shea TP, Williams L, Young S, Nusbaum C, Jaffe DB. ALLPATHS 2: small genomes assembled accurately and with high continuity from short paired reads. Genome Biol, 2009, 10(10): R103.
[67] Schulz MH, Zerbino DR, Vingron M, Birney E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics, 2012, 28(8): 1086-1092.
[68] Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics, 2008, 24(5): 713-714.
[69] Surget-Groba Y, Montoya-Burgos JI. Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res, 2010, 20(10): 1432-1440.
[70] Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury
R, Zeng Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol, 2011, 29(7): 644-652.
[71] Huang XQ, Wang JM, Aluru S, Yang SP, Hillier L. PCAP: A whole-genome assembly program. Genome Res, 2003, 13(9): 2164-2170.
[72] Barbazuk WB, Emrich SJ, Chen HD, Li L, Schnable PS. SNP discoveryvia 454 transcriptome sequencing. Plant J, 2007, 51(5): 910-918
致谢词:
非常感谢老师于百忙之中,在我大学的最后学习阶段,给了我毕业设计很大的帮助,你们放弃了自己的休息时间,你们的这种无私奉献的敬业精神令人钦佩,在此我向你们表示我诚挚的谢意,祝所有的老师培养出越来越多的优秀人才,桃李满天下!