用python进行GEO数据挖掘(学习笔记一):ID转换
在这之前,假设阅读者已经对GEO数据库有了一定的了解,并且知道数据下载的几种方式。需要说明的是,R包GEOquery下载数据对网速要求比较高,假如网速不是很好,推荐用代理服务器“西柚云(网址西柚云生信数据下载代理服务 (xiyoucloud.net))”下载。超快!
进行ID转换之前需要两个文件:
1.基因表达量矩阵文件
2.探针ID与gene symbol对应的文件
 基因矩阵文件,第一列为探针ID,其余列是样本表达量
基因矩阵文件,第一列为探针ID,其余列是样本表达量
 探针ID与gene symbol对应文件
探针ID与gene symbol对应文件
以GSE5281为例
加载pandas库
import pandas as pd读取数据
data_5281=pd.read_csv("F:\\bioinformatics\\20220806\\data\\GSE5281\\data5281.csv")
gene_symbo_5281=pd.read_csv("F:\\bioinformatics\\20220806\\data\\GSE5281\\ids5281.csv")查看数据
print(data_5281)
print(gene_symbo_5281)

可以发现,data_5281中的探针ID与gene symbol文件中探针ID不同,区别在于data_5281中的探针ID有双引号,为了方便后续操作,下面就先去掉双引号
#将两个数据中的探针ID提取出来,转成列表
list1=gene_symbo_5281['prob_id'].tolist()
list2=data_5281['probe_id'].tolist()
##处理列表中多余的字符串
list22=[]
for i in list2:
a=i.replace('"','')
list22.append(a)
##把数据中探针ID多余的双引号删除
data_5281.loc[:,'probe_id']=list22
重新看一下基因表达量矩阵文件data_5281中的探针ID,发现正常了
print(data_5281)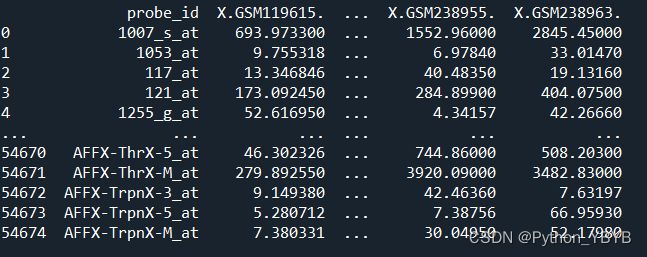
统计表达数据中的探针ID在gene symbol数据中出现的情况
##统计表达数据中的探针ID在ids数据中出现的情况
listx=[]
for j in list22:
if j in list1:
t='y'
else:
t='n'
listx.append(t)
print('TRUE:',listx.count('y'),'FALSE:',listx.count('n')) ##探针id在ids中出现的情况
##运行结果:TRUE: 43138 FALSE: 11537结果显示:有43138个探针ID在gene symbol文件中存在接下来先过滤掉其余的那11537个探针
print('ids中不重复基因的个数:',len(set(gene_symbo_5281['symbol'])))
##运行结果:ids中不重复基因的个数: 20857上述结果说明gene symbol文件中存在一个基因对应多个探针的情况,别急,一步一步地来。
准备提取在ids中存在gene symbol ID的那些数据:
首先找到'y'和'n'与索引的对应,用元组数据(y表示在,n表示不在)
listt=list(enumerate(listx))
print(listt[0:6])
##长成这个样子的:
#[(0, 'y'), (1, 'y'), (2, 'y'), (3, 'y'), (4, 'y'), (5, 'y'),........]其次,提取''y"的下标
##提取存在symbol ID的探针相应的行索引
listsuo=[]
for k in listt:
if k[1]=='y':
listsuo.append(k[0])
print("ids中有探针ID的行索引号:",listsuo)
print('在ids中存在探针ID的元素个数:',len(listsuo)) 上述代码运行结果
上述代码运行结果
也是43138,与之前的数据对应,说明没有问题
最后,删除那些在ids中没有探针ID的11537个探针数据
##删除掉那些在ids中没有探针ID的数据
data_5281_new=data_5281[data_5281.index.isin(listsuo)]
print(data_5281_new)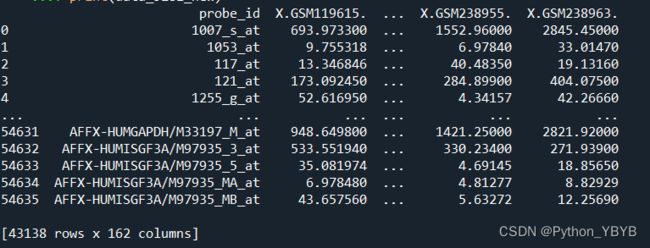
得到的数据维度与预期的一致
接下来就可以进行合并啦
不过好像ids数据中的探针名称是prob_id,与表达矩阵中名称probe_id不一样,先修改一下
gene_symbo_5281_new=gene_symbo_5281.rename(columns={'prob_id':'probe_id'}) #探针名改成一致
print(gene_symbo_5281_new)
现在改过来了就开始进行合并
#合并数据
df=pd.merge(gene_symbo_5281_new,data_5281_new,on='probe_id')
print(df)
到这里有人可能会问,需不需要考虑两个文件中probe_id号的顺序,顺序不一样会不会影响合并结果?
答案是,不会
python中的merge函数与R中的merge函数更优的地方在于,python中的merge函数在以列合并时,不用考虑列中元素的顺序是否一一对应上,merge函数自动以light边列的顺序自动匹配另外一个数据。
上面我们知到symbol id 数据中是一个基因对应多个探针的情况,那么就把同一个symbol ID所对应的多个探针(的行数据)进行分组,为了做到一一对应,计算每一个组中每一个探针在所有样本下的均值,取均值最大的那个探针作为该symbol所对应唯一探针,就OK了!
第一步:计算每一行的均值,并将计算得到的均值添加到新的列
df['values_mean']=df.iloc[:,2:164].apply(lambda x:x.mean(),axis=1)
print(df)
第二步:分组后留下每一个组中均值最大的一个探针
df1=df.groupby('symbol',as_index=False).apply(lambda x:x.nlargest(1,'values_mean'))
可以看到数据维度是20857*164,与预期的结果一致,这样子ID转化就做完了,最后要做的就是删除多余的列就结束!
##删除多于列
df1=pd.DataFrame(df1)
expset=df1.drop(columns=['probe_id','values_mean'])
print(expset)
expset.to_excel('F:\\bioinformatics\\20220806\\data\\GSE5281\\NEW_GSE5281.xlsx',index=False)
161个样本一个symbol ID列共162列,20857个gene symbol ID与之前一致。这就是最后我们要的结果!