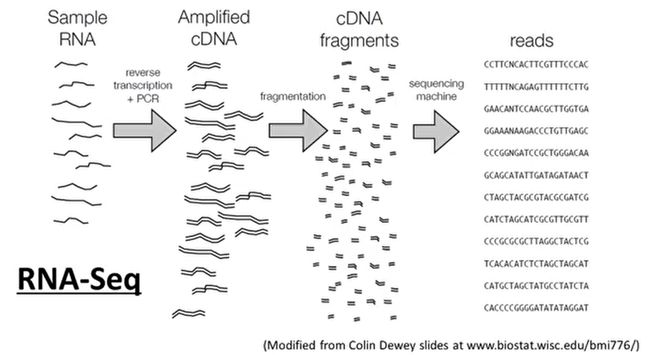
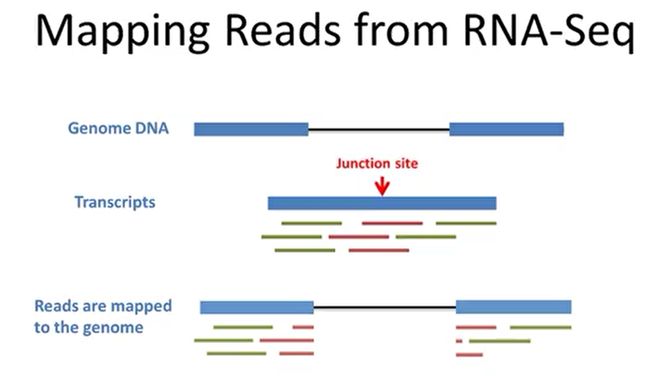
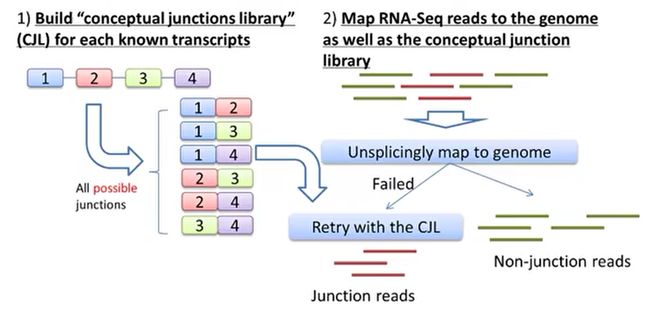
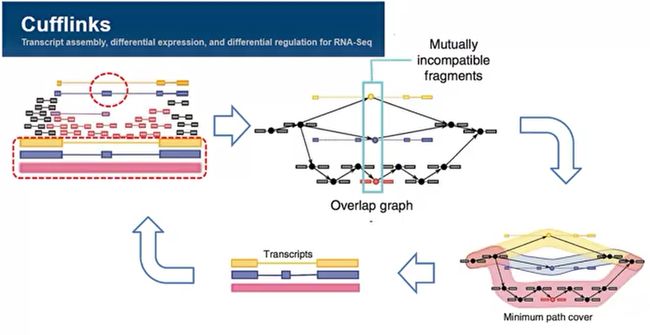
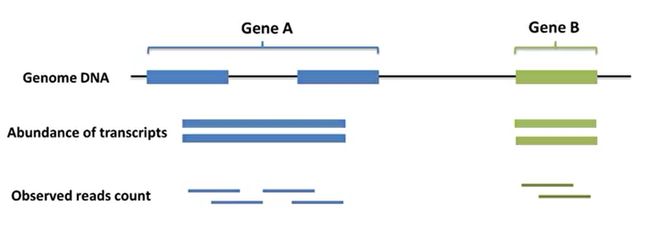
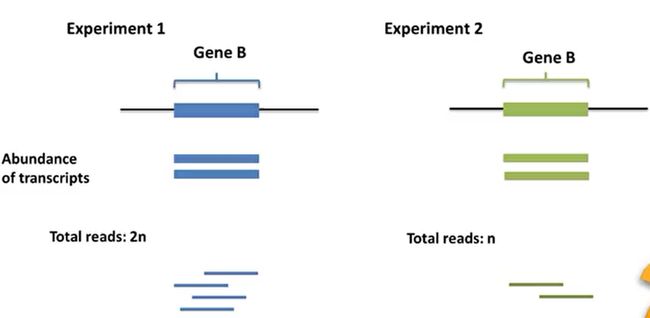
- R语言 关联TCGA数据库下载的RNA-SEQ数据和临床信息
胖胖的胖球
生物信息学大数据生物信息学r语言
刚开始学习TCGA数据处理和分析,记下来方便以后查看setwd("E:/MyData/luadRNA-SEQ-20201028")#把工作目录定位到manifest文件所在的位置manifest="gdc_manifest.2020-10-28.txt"x=read.table(manifest,header=T)#header为TRUE表示读取第一行作为变量名表格已经建好了,可以view(x),
- RNA-seq 差异分析的细节详解 (8)
后端
引言本系列将开展全新的转录组分析专栏,主要针对使用DESeq2时可能出现的问题和方法进行展开描述。想要学习更多内容可以添加文末的学习交流群或客服QQ:941844452。Wald检验的各个步骤DESeq函数依次运行以下函数:dds<-estimateSizeFactors(dds)dds<-estimateDispersions(dds)dds<-nbinomWaldTest(dds)用于估计尺寸
- 单细胞分析(11)——scRNA-seq数据整合
生信小鹏
生信技能学习scRNA单细胞测序经验分享
单细胞RNA-seq数据整合:SeuratIntegrationandHarmony1.研究背景在单细胞RNA测序(scRNA-seq)研究中,批次效应(batcheffect)是不可忽视的问题。不同样本来源(如多个实验室、不同测序平台、不同患者)可能会导致非生物学因素的影响,从而影响数据分析的准确性。之前单独写过Harmony去除批次,为了更好地整合多个样本,这次使用以下两种方法进行批次校正:S
- R中单细胞RNA-seq分析教程 (6)
后端
引言本系列开启R中单细胞RNA-seq数据分析教程,持续更新,欢迎关注,转发!简介现在,很少有人只进行一次单细胞RNA测序实验并仅产生一份数据。原因很直接:目前的单细胞RNA测序技术每次只能捕捉到有限样本的分子状态。为了在多个实验和不同条件下对众多样本进行测量,通常需要对来自不同实验的单细胞RNA测序数据进行联合分析。虽然有些实验策略,比如细胞哈希!,以及一些计算方法,比如demuxlet和scS
- 小麦雌蕊相关基因和网络的共表达网络分析
请你喝好果汁641
文献学习学习
https://peerj.com/articles/13902/#摘要作物雄性不育具有重要的理论研究和育种应用价值。HTS-1的雄蕊转化为雌蕊或雌蕊状结构,是春季三雌蕊(CSTP)小麦中重要的雄性不育材料。然而,HTS-1中雌蕊发育的分子机制仍然是一个谜。11个小麦组织的RNA-seq数据来自美国国家生物技术信息中心(NCBI),包括CSTP的雄蕊和HTS-1的雌蕊和雌蕊。鲑鱼程序用于量化11种
- deseq2进行差异分析时的分组问题
请你喝好果汁641
RNA-seq学习
这段代码是使用DESeq2包进行RNA-Seq数据差异表达分析的示例。它展示了如何在不同实验设计下进行差异表达分析,包括两组比较、两条件两基因型的交互作用,以及两条件三基因型的分析。示例1:两组比较#创建一个示例数据集,包含4个样本dds=1.10中,所选的阈值是过滤器的最低分位数,其中拒绝数接近拟合曲线在过滤器分位数上的峰值。“接近”定义为在1个残差标准差内。对于未通过过滤阈值的基因,调整后的p
- GSEA - Gene set enrichment analysis 基因集富集 | ORA - Over-Representation Analysis 分析原理与应用...
weixin_30294709
python数据库人工智能
R批量做GSEA分析还没有官方的包,但是clusterprofiler可以做,它调用了最新的gfsea包。GeneSetTestingforRNA-seq-fgsea教程RNA-seq是利器,大部分做实验的老板手下都有大量转录组数据,所以RNA-seq的分析需求应该是很大的(大部分的生信从业人员应该都差不多要沾边吧)。普通的转录组套路并不多,差异表达基因、富集分析、WGCNAnetwork以及一些
- 9.单细胞 RNA-seq:聚类分析
denghb001
学习目标:利用多种方法来评估聚类选择的PC基于重要的PC执行单细胞聚类单细胞RNA-seq聚类分析现在我们已经整合了高质量的细胞,我们想知道我们的细胞群中存在的不同细胞类型。image目标:为了生成特定细胞类型的簇,并使用已知的细胞类型的标志基因来确定的簇的身份。为了确定分群是否代表真实的细胞类型或由于生物或技术差异而形成的群集,如在细胞周期的S期的细胞群,特定批次的簇,或具有高线粒体含量的细胞。
- 用DESeq2包来对RNA-seq数据进行差异分析
Seurat_Satija
差异分析的套路都是差不多的,大部分设计思想都是继承limma这个包,DESeq2也不例外。DESeq2是DESeq包的更新版本,看样子应该不会有DESeq3了,哈哈,它的设计思想就是针对count类型的数据。可以是任意features的count数据,比如对各个基因的count,或者外显子,或者CHIP-seq的一些feature,都可以用来做差异分析。使用这个包也是需要三个数据:表达矩阵分组矩阵
- salmon分析RNA-seq实战
超级无敌大蜗牛
Salmon应用查看帮助文档#查看可用的命令###Salmonv0.9.1salmon-h#查看帮助文档之Salmon'squasi-mapping-basedmodesalmon--no-version-checkquant--help-reads#查看帮助文档之Salmon'salignment-basedmodesalmon--no-version-checkquant--help-alig
- RNA-seq数据分析_未完成
子诚之
组学数据分析数据分析
目录基础分析1.质控(reads)2.比对3.质控(alignment)4.定量5.样本合并差异表达1.质控(cohort)2.差异分析3.可视化(差异)富集分析肿瘤免疫1.免疫组库2.免疫浸润3.免疫响应4.新抗原预测微生物组参考本文主要覆盖了肿瘤样本bulkRNA-seq数据常见的分析步骤,并从实践角度出发,较为具体地介绍了每一步骤依赖的工具和数据集。另外,尽管本文适用于肿瘤样本,但其中的一些
- 10X单细胞转录组个性化分析-拟时序分析
Seurat_Satija
在发育过程中,细胞会对刺激做出反应,在整个生命过程中,从一种功能性“状态”转变为另一种功能性“状态”。处于不同状态的细胞表达的基因不同,产生蛋白质和代谢物的动态重复序列,从而完成它们的工作。当细胞在不同状态间转变时,会经历转录重组的过程,其中一些基因被沉默,而另一些基因被激活。这些瞬时状态通常难以表征,因为在更稳定状态之间纯化细胞是困难或不可能的。单细胞RNA-Seq可以使您在不需要纯化细胞的情况
- 转录组结果和qRT-PCR结果又不一致?!
Seurat_
什么?!按照转录组筛选的5个最明显的差异基因只有2个与qRT-PCR结果一致?转录组测序(RNA-seq)将细胞内某一类型(或全部)的RNA逆转录成DNA,通过高通量测序的方法测定其序列并统计其表达水平的一项技术。是检测基因表达变化的通用方法。qRT-PCR是指通过对PCR扩增反应中每一个循环产物荧光信号的实时检测从而实现对起始模板定量及定性的分析。RNA-seq无需知道实验样本的基因组序列含比传
- 11.8 RNA-seq表达rpkm数据操作实践(二)
KK_f2d5
接下来,我们需要对rpkm数据进行annotation再画图。一篇文章说:RPKMvalueswerelog2transformedbeforegeneratingheatmaps.TheheatmapwasgeneratedbyBARHeatmapperPlus(http://bar.utoronto.ca/ntools/cgi-bin/ntools_heatmapper_plus.cgi)on
- 宝藏R包:TCGA的转录组数据挖掘一站搞定
小洁忘了怎么分身
最近在看ceRNA的时候看到了一个宝藏R包,写包简化了芯片数据下游分析之后,我正想着写转录组下游分析的简化版,就看到了它。用起来~0.R包和数据准备if(!require(GDCRNATools))BiocManager::install("GDCRNATools")library(GDCRNATools)这里使用的是作者给的示例数据,RNA-seq是1000行,miRNAseq是2588个。#m
- 生信地基系列--常规分析流程
可能性之兽
还在到底搜索一些R的分析流程吗?biocondutor已经给你准备好了29篇Bioconductor-BiocViewsimage.png注释流程生物导体可以导入多种与序列相关的文件类型,包括Fasta、fastq、BAM、VCF、gff、bed和wig文件等。包支持常见的和高级的序列操作操作,例如修剪、转换和对齐。领域特异性分析包括质量评估、ChIP-seq、差异表达、RNA-seq和其他方法。
- Python版WGCNA分析和蛋白质相互作用PPI分析教程
Starlitnightly
python开发语言
在前面的教程中,我们介绍了使用omicverse完成基本的RNA-seq的分析流程,在本节教程中,我们将介绍如何使用omicverse完成加权基因共表达网络分析WGCNA以及蛋白质相互作用PPI分析。环境的下载在这里我们只需要安装omicverse环境即可,有两个方法:一个是使用conda:condainstallomicverse-cconda-forge另一个是使用pip:pipinstall
- Python版RNA-seq分析教程:DEseq2差异表达基因分析
Starlitnightly
python开发语言
BulkRNA-seq分析的一个重要任务是分析差异表达基因,我们可以用omicverse包来完成这个任务。在omicverse中,除了最简单的ttest外,在这里,我们介绍一种类似R语言中的Deseq2等包的模型来计算差异表达基因。原教程地址:https://omicverse.readthedocs.io/en/latest/Tutorials-bulk/t_deseq2/环境的下载在这里我们只
- python基因差异分析包_一个生信素人的上道经验分享-转录组测序(差异分析篇)...
weixin_39607873
python基因差异分析包
原标题:一个生信素人的上道经验分享-转录组测序(差异分析篇)转录组测序技术(RNA-seq)作为目前二代测序领域最普遍的技术手段,自从转录组测序问世以来,已经开发了数百种分析工具。根据转录组分析内容可大致将其分析流程分为比对,转录本组装,基因注释和差异表达分析。目前,分析的每一步都有很多软件,其软件的性能和分析效率不尽相同。上篇文章小编为各位小伙伴介绍了转录组分析的第三步——基因定量【一个生信素人
- SLICER:从单细胞RNA-seq数据推断分支的非线性细胞轨迹
生信编程日常
image.pngSLICER是一种构建轨迹的算法,该轨迹描述了某些生物学过程中基因表达的变化。SLICER可以捕获高度非线性的基因表达变化,自动选择与该过程相关的基因,并检测轨迹中的多个分支和loopfeatures。SLICER(SelectiveLocallyLinearInferenceofCellularExpressionRelationships),是一种使用局部线性嵌入(LLE)重
- 10.单细胞 RNA-seq:聚类分析
denghb001
学习目标:评估是否存在聚类过程产生的技术误差使用PCA和UMAP图确定聚类质量,并了解何时重新聚类评估已知的细胞类型标记与假设簇的细胞类型同一性单细胞RNA-seq聚类分析现在我们已经进行了整合,我们想知道我们的细胞群中存在哪些不同细胞类型。image目标:*生成特定于细胞类型的簇,并使用已知的标记来确定簇的身份。确定分群是否代表真实的细胞类型或由于生物或技术差异而形成的群集,例如处于细胞周期S期
- 新版EvidenceModeler基因组注释方法
在学生信秃头中
EvidenceModeler用于将多种方法的注释结果合并整理从头预测的结果,同源注释结果,RNA-seq辅助注释结果,EST注释结果等等。1.下载并配置环境wgethttps://github.com/EVidenceModeler/EVidenceModeler/releases/download/EVidenceModeler-v2.1.0/EVidenceModeler-v2.1.0.ta
- 小鼠嗅球的单细胞rna-seq揭示了细胞的异质性和成体神经元活性依赖的分子普查
猫姐Lily
Single-CellRNA-SeqofMouseOlfactoryBulbRevealsCellularHeterogeneityandActivity-DependentMolecularCensusofAdult-BornNeurons题目:小鼠嗅球的单细胞rna-seq揭示了细胞的异质性和成体神经元活性依赖的分子普查作者及单位:BurakTepe,MatthewC.Hill,Brandon
- 看了老大比较基因组学视频后扩展的小内容
小梦游仙境
老大视频比较基因组在IGV看了http://www.bio-info-trainee.com/2218.htmlwes:探针捕获rna-seq:不会出现外显子两边下降chip-seq:不会出现整齐,得到的文件是peaksbedfilesRNA-seq这十年https://mp.weixin.qq.com/s/a3y46NNNO-wardO3XWwh0w经过不断的技术开发和改进,以Roche公司的4
- STAR: ultrafast universal RNA-seq aligner
sunlight_yy
DobinA,DavisCA,SchlesingerF,etal.STAR:ultrafastuniversalRNA-seqaligner[J].Bioinformatics,2012,29(1).ABSTRACTMotivation:高通量RNA-seq数据的准确比对是一个具有挑战性但尚未解决的问题,因为转录结构不连续,读取长度相对较短且测序技术的通量不断提高。当前可用的RNA-seq比对仪遭
- 学习:StatQuest-RNA-seq技术重复
小潤澤
概念首先我们先辨析一下生物学重复和技术重复,我们的RNA-seq的data产生差异是正常的,关于差异来自于几个方面:生物学多样性,个体间差异,即便是同一物种(基因组相同),其转录本也未必都相同image.png对于geneX来说,不同的小鼠其表达量都不同image.png计算其平均值如上,其中μ是平均值技术多样性,即便是对于同一物种,两次测定的结果都不相同image.png绿色箭头表示技术重复带来
- 2022-11-29 RNA-seq差异表达分析
Zheng_xy
最近看到一篇文章提到当样本量很大的时候差异表达分析使用秩和检验效果较好。文章题目:ExaggeratedfalsepositivesbypopulardifferentialexpressionmethodswhenanalyzinghumanpopulationsamplesGenomeBiol.2022Mar15;23(1):79.doi:10.1186/s13059-022-02648-4.
- 2022-08-14
佳奥
上一次写随笔还是两周前八月第一周在玩异度之刃3.....第二周把EGO数据库挖掘的内容结束了,另外学了chip-seq全流程,这一次感觉比rna-seq那一趟顺利多了,当然,数据量也是大多了,分析结束后还要把WSL占用的空间重新释放出来,属实头大。chip-seq实战九、十已经完成,每天更新一篇。假期还剩两周左右,接下来学什么呢?我想完善一下,会使用Linux,会使用R语言,学了转录组分析的流程和
- 学习:StatQuest-Heatmap
小潤澤
Heatmapimage.png在RNA-seq中热图往往用于衡量不同样本不同基因的表达情况(主要看上下表达),这个图就是个热图,横坐标表示不同样本,纵坐标表示基因。热图中的标准化和聚类Z-scoreimage.png如果有一列数据,我们要计算Z-score:计算这组数据的均值每个数据点减去均值计算标准差用第二步计算的值除以标准差image.png标准化有对某一基因标准化的,有对每个样本进行标准化
- RNA-seq转录组数据分析
医学小白学生信
B站:RNA-seq转录组数据分析入门实战1linux常用命令touchtext.txt#新建文件rm-rf/var/log/httpd/access#将会删除/var/log/httpd/access目录以及其下所有文件、文件夹rm-f*html#删除所有html格式文件rm-f*zip#删除所有zip格式文件tarzxvf#解压tar.gz文件tarjxvfsamtools-1.11.tar.
- java责任链模式
3213213333332132
java责任链模式村民告县长
责任链模式,通常就是一个请求从最低级开始往上层层的请求,当在某一层满足条件时,请求将被处理,当请求到最高层仍未满足时,则请求不会被处理。
就是一个请求在这个链条的责任范围内,会被相应的处理,如果超出链条的责任范围外,请求不会被相应的处理。
下面代码模拟这样的效果:
创建一个政府抽象类,方便所有的具体政府部门继承它。
package 责任链模式;
/**
*
- linux、mysql、nginx、tomcat 性能参数优化
ronin47
一、linux 系统内核参数
/etc/sysctl.conf文件常用参数 net.core.netdev_max_backlog = 32768 #允许送到队列的数据包的最大数目
net.core.rmem_max = 8388608 #SOCKET读缓存区大小
net.core.wmem_max = 8388608 #SOCKET写缓存区大
- php命令行界面
dcj3sjt126com
PHPcli
常用选项
php -v
php -i PHP安装的有关信息
php -h 访问帮助文件
php -m 列出编译到当前PHP安装的所有模块
执行一段代码
php -r 'echo "hello, world!";'
php -r 'echo "Hello, World!\n";'
php -r '$ts = filemtime("
- Filter&Session
171815164
session
Filter
HttpServletRequest requ = (HttpServletRequest) req;
HttpSession session = requ.getSession();
if (session.getAttribute("admin") == null) {
PrintWriter out = res.ge
- 连接池与Spring,Hibernate结合
g21121
Hibernate
前几篇关于Java连接池的介绍都是基于Java应用的,而我们常用的场景是与Spring和ORM框架结合,下面就利用实例学习一下这方面的配置。
1.下载相关内容: &nb
- [简单]mybatis判断数字类型
53873039oycg
mybatis
昨天同事反馈mybatis保存不了int类型的属性,一直报错,错误信息如下:
Caused by: java.lang.NumberFormatException: For input string: "null"
at sun.mis
- 项目启动时或者启动后ava.lang.OutOfMemoryError: PermGen space
程序员是怎么炼成的
eclipsejvmtomcatcatalina.sheclipse.ini
在启动比较大的项目时,因为存在大量的jsp页面,所以在编译的时候会生成很多的.class文件,.class文件是都会被加载到jvm的方法区中,如果要加载的class文件很多,就会出现方法区溢出异常 java.lang.OutOfMemoryError: PermGen space.
解决办法是点击eclipse里的tomcat,在
- 我的crm小结
aijuans
crm
各种原因吧,crm今天才完了。主要是接触了几个新技术:
Struts2、poi、ibatis这几个都是以前的项目中用过的。
Jsf、tapestry是这次新接触的,都是界面层的框架,用起来也不难。思路和struts不太一样,传说比较简单方便。不过个人感觉还是struts用着顺手啊,当然springmvc也很顺手,不知道是因为习惯还是什么。jsf和tapestry应用的时候需要知道他们的标签、主
- spring里配置使用hibernate的二级缓存几步
antonyup_2006
javaspringHibernatexmlcache
.在spring的配置文件中 applicationContent.xml,hibernate部分加入
xml 代码
<prop key="hibernate.cache.provider_class">org.hibernate.cache.EhCacheProvider</prop>
<prop key="hi
- JAVA基础面试题
百合不是茶
抽象实现接口String类接口继承抽象类继承实体类自定义异常
/* * 栈(stack):主要保存基本类型(或者叫内置类型)(char、byte、short、 *int、long、 float、double、boolean)和对象的引用,数据可以共享,速度仅次于 * 寄存器(register),快于堆。堆(heap):用于存储对象。 */ &
- 让sqlmap文件 "继承" 起来
bijian1013
javaibatissqlmap
多个项目中使用ibatis , 和数据库表对应的 sqlmap文件(增删改查等基本语句),dao, pojo 都是由工具自动生成的, 现在将这些自动生成的文件放在一个单独的工程中,其它项目工程中通过jar包来引用 ,并通过"继承"为基础的sqlmap文件,dao,pojo 添加新的方法来满足项
- 精通Oracle10编程SQL(13)开发触发器
bijian1013
oracle数据库plsql
/*
*开发触发器
*/
--得到日期是周几
select to_char(sysdate+4,'DY','nls_date_language=AMERICAN') from dual;
select to_char(sysdate,'DY','nls_date_language=AMERICAN') from dual;
--建立BEFORE语句触发器
CREATE O
- 【EhCache三】EhCache查询
bit1129
ehcache
本文介绍EhCache查询缓存中数据,EhCache提供了类似Hibernate的查询API,可以按照给定的条件进行查询。
要对EhCache进行查询,需要在ehcache.xml中设定要查询的属性
数据准备
@Before
public void setUp() {
//加载EhCache配置文件
Inpu
- CXF框架入门实例
白糖_
springWeb框架webserviceservlet
CXF是apache旗下的开源框架,由Celtix + XFire这两门经典的框架合成,是一套非常流行的web service框架。
它提供了JAX-WS的全面支持,并且可以根据实际项目的需要,采用代码优先(Code First)或者 WSDL 优先(WSDL First)来轻松地实现 Web Services 的发布和使用,同时它能与spring进行完美结合。
在apache cxf官网提供
- angular.equals
boyitech
AngularJSAngularJS APIAnguarJS 中文APIangular.equals
angular.equals
描述:
比较两个值或者两个对象是不是 相等。还支持值的类型,正则表达式和数组的比较。 两个值或对象被认为是 相等的前提条件是以下的情况至少能满足一项:
两个值或者对象能通过=== (恒等) 的比较
两个值或者对象是同样类型,并且他们的属性都能通过angular
- java-腾讯暑期实习生-输入一个数组A[1,2,...n],求输入B,使得数组B中的第i个数字B[i]=A[0]*A[1]*...*A[i-1]*A[i+1]
bylijinnan
java
这道题的具体思路请参看 何海涛的微博:http://weibo.com/zhedahht
import java.math.BigInteger;
import java.util.Arrays;
public class CreateBFromATencent {
/**
* 题目:输入一个数组A[1,2,...n],求输入B,使得数组B中的第i个数字B[i]=A
- FastDFS 的安装和配置 修订版
Chen.H
linuxfastDFS分布式文件系统
FastDFS Home:http://code.google.com/p/fastdfs/
1. 安装
http://code.google.com/p/fastdfs/wiki/Setup http://hi.baidu.com/leolance/blog/item/3c273327978ae55f93580703.html
安装libevent (对libevent的版本要求为1.4.
- [强人工智能]拓扑扫描与自适应构造器
comsci
人工智能
当我们面对一个有限拓扑网络的时候,在对已知的拓扑结构进行分析之后,发现在连通点之后,还存在若干个子网络,且这些网络的结构是未知的,数据库中并未存在这些网络的拓扑结构数据....这个时候,我们该怎么办呢?
那么,现在我们必须设计新的模块和代码包来处理上面的问题
- oracle merge into的用法
daizj
oraclesqlmerget into
Oracle中merge into的使用
http://blog.csdn.net/yuzhic/article/details/1896878
http://blog.csdn.net/macle2010/article/details/5980965
该命令使用一条语句从一个或者多个数据源中完成对表的更新和插入数据. ORACLE 9i 中,使用此命令必须同时指定UPDATE 和INSE
- 不适合使用Hadoop的场景
datamachine
hadoop
转自:http://dev.yesky.com/296/35381296.shtml。
Hadoop通常被认定是能够帮助你解决所有问题的唯一方案。 当人们提到“大数据”或是“数据分析”等相关问题的时候,会听到脱口而出的回答:Hadoop! 实际上Hadoop被设计和建造出来,是用来解决一系列特定问题的。对某些问题来说,Hadoop至多算是一个不好的选择,对另一些问题来说,选择Ha
- YII findAll的用法
dcj3sjt126com
yii
看文档比较糊涂,其实挺简单的:
$predictions=Prediction::model()->findAll("uid=:uid",array(":uid"=>10));
第一个参数是选择条件:”uid=10″。其中:uid是一个占位符,在后面的array(“:uid”=>10)对齐进行了赋值;
更完善的查询需要
- vim 常用 NERDTree 快捷键
dcj3sjt126com
vim
下面给大家整理了一些vim NERDTree的常用快捷键了,这里几乎包括了所有的快捷键了,希望文章对各位会带来帮助。
切换工作台和目录
ctrl + w + h 光标 focus 左侧树形目录ctrl + w + l 光标 focus 右侧文件显示窗口ctrl + w + w 光标自动在左右侧窗口切换ctrl + w + r 移动当前窗口的布局位置
o 在已有窗口中打开文件、目录或书签,并跳
- Java把目录下的文件打印出来
蕃薯耀
列出目录下的文件文件夹下面的文件目录下的文件
Java把目录下的文件打印出来
>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>>
蕃薯耀 2015年7月11日 11:02:
- linux远程桌面----VNCServer与rdesktop
hanqunfeng
Desktop
windows远程桌面到linux,需要在linux上安装vncserver,并开启vnc服务,同时需要在windows下使用vnc-viewer访问Linux。vncserver同时支持linux远程桌面到linux。
linux远程桌面到windows,需要在linux上安装rdesktop,同时开启windows的远程桌面访问。
下面分别介绍,以windo
- guava中的join和split功能
jackyrong
java
guava库中,包含了很好的join和split的功能,例子如下:
1) 将LIST转换为使用字符串连接的字符串
List<String> names = Lists.newArrayList("John", "Jane", "Adam", "Tom");
- Web开发技术十年发展历程
lampcy
androidWeb浏览器html5
回顾web开发技术这十年发展历程:
Ajax
03年的时候我上六年级,那时候网吧刚在小县城的角落萌生。传奇,大话西游第一代网游一时风靡。我抱着试一试的心态给了网吧老板两块钱想申请个号玩玩,然后接下来的一个小时我一直在,注,册,账,号。
彼时网吧用的512k的带宽,注册的时候,填了一堆信息,提交,页面跳转,嘣,”您填写的信息有误,请重填”。然后跳转回注册页面,以此循环。我现在时常想,如果当时a
- 架构师之mima-----------------mina的非NIO控制IOBuffer(说得比较好)
nannan408
buffer
1.前言。
如题。
2.代码。
IoService
IoService是一个接口,有两种实现:IoAcceptor和IoConnector;其中IoAcceptor是针对Server端的实现,IoConnector是针对Client端的实现;IoService的职责包括:
1、监听器管理
2、IoHandler
3、IoSession
- ORA-00054:resource busy and acquire with NOWAIT specified
Everyday都不同
oraclesessionLock
[Oracle]
今天对一个数据量很大的表进行操作时,出现如题所示的异常。此时表明数据库的事务处于“忙”的状态,而且被lock了,所以必须先关闭占用的session。
step1,查看被lock的session:
select t2.username, t2.sid, t2.serial#, t2.logon_time
from v$locked_obj
- javascript学习笔记
tntxia
JavaScript
javascript里面有6种基本类型的值:number、string、boolean、object、function和undefined。number:就是数字值,包括整数、小数、NaN、正负无穷。string:字符串类型、单双引号引起来的内容。boolean:true、false object:表示所有的javascript对象,不用多说function:我们熟悉的方法,也就是
- Java enum的用法详解
xieke90
enum枚举
Java中枚举实现的分析:
示例:
public static enum SEVERITY{
INFO,WARN,ERROR
}
enum很像特殊的class,实际上enum声明定义的类型就是一个类。 而这些类都是类库中Enum类的子类 (java.l