单细胞测序数据整合
一、单细胞测序数据整合的原理
单细胞测序数据的整合,有点类似于基因组的拼接和比对过程。整合过程需要找到两个dataset之间的相似的部分,在单细胞测序数据则意味着两个两次不同实验的dataset中有一部分细胞具有相似的生物学状态(虽然这簇细胞的基因表达绝对值不一定相同,但是这簇细胞整体具有一致性或相似性)。

A 两个来自不同实验的单细胞数据,有相似生物学状态的细胞群,但是query dataset具有特有的细胞群
B 进行常规的相关性分析,并进行Log2标准化处理。
C 在同一个共享空间下,鉴定两个dataset之间相互最近的邻居(MNN),这些cells就可以作为两个dataset之间的anchors细胞,从而帮助进行dataset 整合。
D 对于每一个anchor对,会给予一致性给出打分值
E 基于这些anchors细胞以及打分值,计算矫正向量,从而进行数据集的整合
来源:单细胞测序数据整合 - 知乎 (zhihu.com) https://zhuanlan.zhihu.com/p/158974557?ivk_sa=1024320u
https://zhuanlan.zhihu.com/p/158974557?ivk_sa=1024320u
二、一般步骤
来源:单细胞数据整合 Comprehensive Integration - 知乎 (zhihu.com)https://zhuanlan.zhihu.com/p/465227964
(1)Normalization
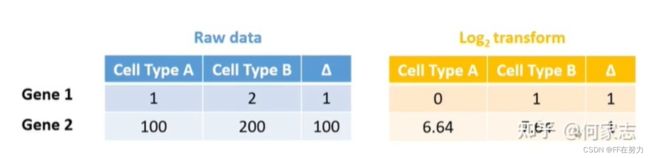
在所有的分析中,我们都要对 single-cell RNA-seq数据集采用标准的预处理。除非特别声明,我们首先对所有的数据集进行 log-normalization, lognormalization 的目的 是防止数据差异完全被高度表达的基因控制,在log-normalization 后, 它就会与基因表达水平独立, 使得表达水平较低的基因不易被忽视
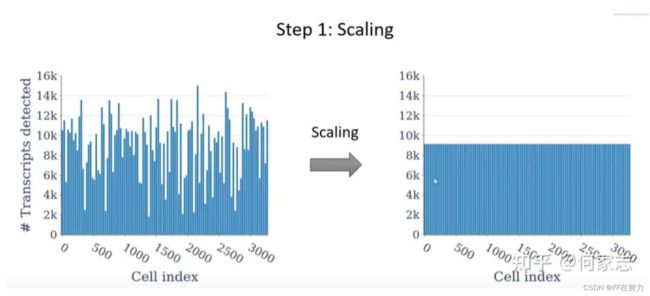
之后,我们再做 z-score transformation, 它是进行降维操作如PCA之前 的一个标准步骤。z-scroe transformation 的目的见下图,由于技术的原因,不同细胞检测到的转录水平是不一样的,即数据有noise, 进过 transfrom之后,不同细胞之间的数据才有可比性。
(2)Feature selection for individual datasets
在每个数据集中,我们接下来要进行特征选择,选择出一组特征(e.g. 基因) ,使得它们在不同的细胞间展现出高度的变化,可以首先用于下游分析。
对每个基因,我们计算它在所有细胞中的标准值的方差,我们直接使用这个方差对特征排序,选取2000个有最大标准方差的基因 作为“高变基因”。 这一过程可以用Seurat v3中的FindVariableFeatures 函数实现。
(3) Feature selection for integrated analysis of multiple datasets
在整合所有数据集的时候,我们要给高变基因优先权。因此, 我们首先单独对每个数据集特征选择,使用上面说的流程。然后看每个基因在所有的数据集中单独被认定为高变基因的个数,用这个指标来作排序,选取top 2000的基因用作 下游分析。 这些步骤可以用Seurat v3中的函数 SelectIntegrationFeatures 来实现。
(4)Identification of anchor correspondences between two datasets
整合分析的关键步骤是找出两个数据集的 锚点(anchors)。锚点代表两个细胞(每个数据集各一个细胞),它们是相关性最大的一对来自不同数据集的细胞,我们预测它们来自一个共同的生物状态。因此找到了锚点,我们就能判断不同数据集间的细胞关系,从而有利于后续的整合操作。 Anchors for reference assembly or transfer learning 的计算可以分别使用Seurat v3包中的函数 FindIntegrationAnchors 和 FindTransferAnchors 。
三、代码实现
方法一:Seurat v3的标准整合流程
(1)数据获取
#清空环境,养成好习惯
rm(list = ls())
setwd("")
library(Seurat)
library(multtest)
library(dplyr)
library(ggplot2)
library(patchwork)
library(SeuratData)#涵盖函数InstallData和LoadData
library(tidyverse)测试数据选择Seurat提供的ifnb数据集,其中包含两个PBMC样品,一个为干扰素刺激处理,另一个为对照。
#install.packages('devtools')
library(devtools)
devtools::install_github('satijalab/seurat-data',force = TRUE)
InstallData("ifnb")
LoadData("ifnb")将ifnb根据stim状态拆分为两个list (stim and CTRL),其中包含两个PBMC样品,一个为干扰素刺激处理,另一个为对照
ifnb.list <- SplitObject(ifnb, split.by = "stim")
C57 <- ifnb.list$CTRL
AS1 <- ifnb.list$STIM(2)数据处理
分别进行数据标准化+选择高变基因
myfunction1 <- function(testA.seu){
testA.seu <- NormalizeData(testA.seu, normalization.method = "LogNormalize", scale.factor = 10000)
testA.seu <- FindVariableFeatures(testA.seu, selection.method = "vst", nfeatures = 2000)
return(testA.seu)
}
C57 <- myfunction1(C57)
AS1 <- myfunction1(AS1)Integration ----FindIntegrationAnchors用来寻找样本整合的数据点(anchors)
testAB.anchors <- FindIntegrationAnchors(object.list = list(C57,AS1), dims = 1:20)
testAB.integrated <- IntegrateData(anchorset = testAB.anchors, dims = 1:20)需要注意的是:上面的整合步骤相对于harmony整合方法,对于较大的数据集(几万个细胞)非常消耗内存和时间,大约9G的数据32G的内存就已经无法运行;当存在某一个Seurat对象细胞数很少(印象中200以下这样子),会报错,这时建议用harmony整合方法
(3)数据分析及可视化
1.切换到整合后的assay,设置为integrated可以理解为:这是整合后的数据。您可以将其看作批处理调整的数据。接下来的分析将基于这种整合数据
DfaultAssay(testAB.integrated) <- "integrated"
# # Run the standard workflow for visualization and clustering
testAB.integrated <- ScaleData(testAB.integrated, features = rownames(testAB.integrated))
testAB.integrated <- RunPCA(testAB.integrated, npcs = 50, verbose = FALSE)
testAB.integrated <- FindNeighbors(testAB.integrated, dims = 1:30)
testAB.integrated <- FindClusters(testAB.integrated, resolution = 0.5)
testAB.integrated <- RunUMAP(testAB.integrated, dims = 1:30)
testAB.integrated <- RunTSNE(testAB.integrated, dims = 1:30)
p1<- DimPlot(testAB.integrated,label = T,split.by = 'stim')#integrated

# Visualization
p2 <- DimPlot(immune.combined, reduction = "umap", group.by = "stim")
p3 <- DimPlot(immune.combined, reduction = "umap", label = TRUE, repel = TRUE)


2.将DefaultAssay设置为“RNA”,意味着接下来的分析将基于原始值,进行识别保守细胞类型标记,可以使用整合的数据进行聚类。但是对差异基因使用integrated是不合适的,而且大多数工具只接受原始的DE计数。
DefaultAssay(testAB.integrated) <- "RNA"
testAB.integrated <- ScaleData(testAB.integrated, features = rownames(testAB.integrated))
testAB.integrated <- RunPCA(testAB.integrated, npcs = 50, verbose = FALSE)
testAB.integrated <- FindNeighbors(testAB.integrated, dims = 1:30)
testAB.integrated <- FindClusters(testAB.integrated, resolution = 0.5)
testAB.integrated <- RunUMAP(testAB.integrated, dims = 1:30)
testAB.integrated <- RunTSNE(testAB.integrated, dims = 1:30)
p4 <- DimPlot(testAB.integrated,label = T,split.by = 'stim')
p1|p4 
可以看到以原始值为分析对象的话,会出现批次效应,两个样本几乎没有重叠的细胞群,Integration方法可以去除批次效应,这些方法首先识别处于匹配生物学状态(anchors)的跨数据集细胞对,然后基于这些anchors校正数据集之间的批次效应。测试样品经过批次矫正后,相同类型的细胞都聚到了一起,这样更有利于对细胞类型进行鉴定。
方法二、harmony的方法
主要优点是:速度快,内存小
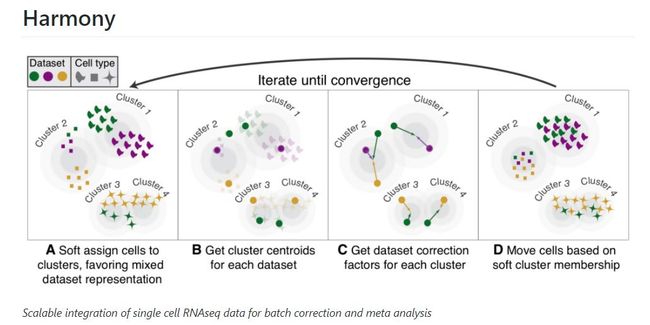
基本原理:我们用不同颜色表示不同数据集,用形状表示不同的细胞类型。首先,Harmony应用主成分分析将转录组表达谱嵌入到低维空间中,然后应用迭代过程去除数据集特有的影响。
(A)Harmony概率性地将细胞分配给cluster,从而使每个cluster内数据集的多样性最大化。 (B)Harmony计算每个cluster的所有数据集的全局中心,以及特定数据集的中心。
(C)在每个cluster中,Harmony基于中心为每个数据集计算校正因子。
(D)最后,Harmony使用基于C的特定于细胞的因子校正每个细胞。由于Harmony使用软聚类,因此可以通过多个因子的线性组合对其A中进行的软聚类分配进行线性校正,来修正每个单细胞。 重复步骤A到D,直到收敛为止。聚类分配和数据集之间的依赖性随着每一轮的减少而减小。
来源:“harmony”整合不同平台的单细胞数据之旅 - 云+社区 - 腾讯云 (tencent.com)https://cloud.tencent.com/developer/article/1650648
1.数据处理
在运行Harmony之前,创建一个Seurat对象并按照标准PCA进行分析,流程同第一个方法
if(!require(harmony))devtools::install_github("immunogenomics/harmony")
test.seu <- pbmc
test.seu <- test.seu%>%
Seurat::NormalizeData() %>%
FindVariableFeatures(selection.method = "vst", nfeatures = 2000) %>%
ScaleData()
test.seu <- RunPCA(test.seu, npcs = 50, verbose = FALSE)2.Run Harmony
运行Harmony的最简单方法是传递Seurat对象并指定要集成的变量。RunHarmony返回Seurat对象,并使用更正后的Harmony坐标。让我们将plot_convergence设置为TRUE,这样我们就可以确保Harmony目标函数在每一轮中都变得更好。

#####先运⾏Seurat标准流程到PCA这⼀步,然后就是Harmony整合,可以简单把这⼀步理解为⼀种新的降维########
test.seu=test.seu %>% RunHarmony("stim", plot_convergence = TRUE)
#设置reduction = 'harmony',后续分析就会调用Harmony矫正之后的PCA降维数据。
test.seu <- test.seu %>%
RunUMAP(reduction = "harmony", dims = 1:30) %>%
FindNeighbors(reduction = "harmony", dims = 1:30) %>%
FindClusters(resolution = 0.5) %>%
identity()
test.seu <- test.seu %>%
RunTSNE(reduction = "harmony", dims = 1:30)
3.可视化
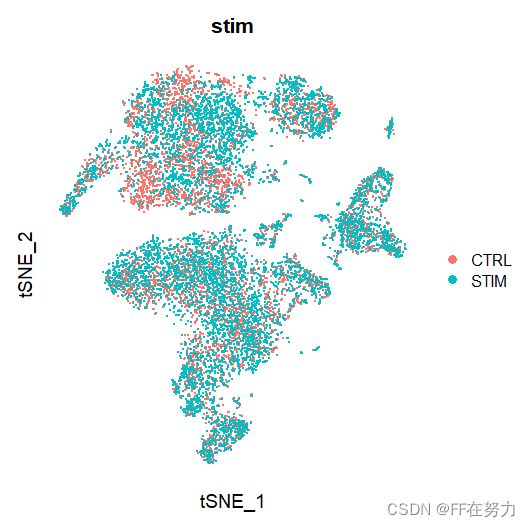
#按照合并的分组进行tsne作图,group.by = "stim"
p5 <- DimPlot(test.seu, reduction = "tsne", group.by = "stim", pt.size=0.5)+theme(
axis.line = element_blank(),
axis.ticks = element_blank(),axis.text = element_blank()
)
#根据细胞类型进行tsne作图,group.by = "ident"
p6 <- DimPlot(test.seu, reduction = "tsne", group.by = "ident", pt.size=0.5, label = TRUE,repel = TRUE)+theme(
axis.line = element_blank(),
axis.ticks = element_blank(),axis.text = element_blank()
)