偏最小二乘法 Partial Least Squares
本文前部分摘自:偏最小二乘法回归(Partial Least Squares Regression),后半部分原创。
诸如基因组学、转录组学、蛋白组学及代谢组学等高通量数据分析,由于自变量数目大于病例数(未知数大于方程个数),无法直接使用传统的统计分析模型。比如,线性回归的窘境:如果样例数m相比特征数n少(m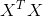 不可逆)。因此最小二乘法
不可逆)。因此最小二乘法![]() 就会失效。遇到这种情况,需要先降维处理。有监督的降维方法,除了常用的LASSO,还有一种叫PLS(偏最小二乘法 Partial Least Squares,或称PLSR 偏最小二乘法回归)。PLSR感觉已经把成分分析和回归发挥到极致了。本文将介绍PLS的原理和应用。
就会失效。遇到这种情况,需要先降维处理。有监督的降维方法,除了常用的LASSO,还有一种叫PLS(偏最小二乘法 Partial Least Squares,或称PLSR 偏最小二乘法回归)。PLSR感觉已经把成分分析和回归发挥到极致了。本文将介绍PLS的原理和应用。
主成分分析 PCA
本部分摘自:偏最小二乘法回归(Partial Least Squares Regression)
先回顾下PCA。令X表示样本,含有m个样例![]() ,每个样例特征维度为n,
,每个样例特征维度为n,![]() 。假设我们已经做了每个特征均值为0处理。
。假设我们已经做了每个特征均值为0处理。
如果X的秩小于n,那么X的协方差矩阵 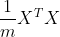 的秩小于n,因此直接使用线性回归的话不能使用最小二乘法来求解出唯一的
的秩小于n,因此直接使用线性回归的话不能使用最小二乘法来求解出唯一的 ,我们想使用PCA来使得可逆,这样就可以用最小二乘法来进行回归了,这样的回归称为主元回归(PCR)。
,我们想使用PCA来使得可逆,这样就可以用最小二乘法来进行回归了,这样的回归称为主元回归(PCR)。
PCA的一种表示形式:T=XP
其中X是样本矩阵,P是X的协方差矩阵的特征向量(当然是按照特征值排序后选取的前r个特征向量),T是X在由P形成的新的正交子空间上的投影(也是样本X降维后的新矩阵)。
PCA的另一种表示:
![]() (假设X秩为n)
(假设X秩为n)
这个公式其实和上面的表示方式T=XP没什么区别。
![]() (当然我们认为P是n*n的,因此
(当然我们认为P是n*n的,因此![]() )
)
如果P是n*r的,也就是舍弃了特征值较小的特征向量,那么上面的加法式子就变成了
这里的E是残差矩阵。其实这个式子有着很强的几何意义, 是第
是第  大特征值对应的归一化后的特征向量,
大特征值对应的归一化后的特征向量, 就是X在上的投影。
就是X在上的投影。![]() 就是X先投影到上,还以原始坐标系得到的X’。下面这个图可以帮助理解:
就是X先投影到上,还以原始坐标系得到的X’。下面这个图可以帮助理解:

黑色线条表示原始坐标系,蓝色的点是原始的4个2维的样本点,做完PCA后,得到两个正交的特征向量坐标 和
和 。绿色点是样本点在上的投影(具有最大方差),红色点是在上的投影。
。绿色点是样本点在上的投影(具有最大方差),红色点是在上的投影。 的每个分量是绿色点在上的截距,
的每个分量是绿色点在上的截距,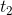 是红色点在上的截距。
是红色点在上的截距。![]() 中的每个分量都可以看做是方向为,截距为相应分量大小的向量,如那个上的橘色箭头。
中的每个分量都可以看做是方向为,截距为相应分量大小的向量,如那个上的橘色箭头。![]() 就得到了X在的所有投影向量,由于和正交,因此
就得到了X在的所有投影向量,由于和正交,因此![]() 就相当于每个点的橘色箭头的加和,可想而知,得到了原始样本点。
就相当于每个点的橘色箭头的加和,可想而知,得到了原始样本点。
如果舍弃了一些特征向量如,那么通过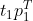 只能还原出原始点的部分信息(得到的绿色点,丢失了蓝色点在另一维度上的信息)。另外,P有个名字叫做loading矩阵,T叫做score矩阵。
只能还原出原始点的部分信息(得到的绿色点,丢失了蓝色点在另一维度上的信息)。另外,P有个名字叫做loading矩阵,T叫做score矩阵。
PLSR思想及步骤
本部分摘自:偏最小二乘法回归(Partial Least Squares Regression)
我们还需要回味一下CCA来引出PLSR。在CCA中,我们将X和Y分别投影到直线得到u和v,然后计算u和v的Pearson系数(也就是Corr(u,v)),认为相关度越大越好。形式化表示:

其中a和b就是要求的投影方向。
想想CCA的缺点:对特征的处理方式比较粗糙,用的是线性回归来表示u和x的关系,u也是x在某条线上的投影,因此会存在线性回归的一些缺点。我们想把PCA的成分提取技术引入CCA,使得u和v尽可能携带样本的最主要信息。还有一个更重要的问题,CCA是寻找X和Y投影后u和v的关系,显然不能通过该关系来还原出X和Y,也就是找不到X到Y的直接映射。这也是使用CCA预测时大多配上KNN的原因。
而PLSR更加聪明,同时兼顾PCA和CCA,并且解决了X和Y的映射问题。看PCA Revisited的那张图,假设对于CCA,X的投影直线是,那么CCA只考虑了X的绿色点与Y在某条直线上投影结果的相关性,丢弃了X和Y在其他维度上的信息,因此不存在X和Y的映射。而PLSR会在CCA的基础上再做一步,由于原始蓝色点可以认为是绿色点和红色点的叠加,因此先使用X的绿色点对Y做回归( ![]() ,样子有点怪,两边都乘以
,样子有点怪,两边都乘以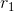 就明白了,这里的Y类似于线性回归里的
就明白了,这里的Y类似于线性回归里的 ,类似
,类似 ),然后用X的红色点对Y的剩余部分F做回归( 得到
),然后用X的红色点对Y的剩余部分F做回归( 得到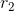 , )。这样Y就是两部分回归的叠加。当新来一个x时,投影一下得到其绿色点和红色点,然后通过r就可以还原出Y,实现了X到Y的映射。当然这只是几何上的思想描述,跟下面的细节有些出入。
, )。这样Y就是两部分回归的叠加。当新来一个x时,投影一下得到其绿色点和红色点,然后通过r就可以还原出Y,实现了X到Y的映射。当然这只是几何上的思想描述,跟下面的细节有些出入。
下面正式介绍PLSR:
1) 设X和Y都已经过标准化(包括减均值、除标准差等)。
2) 设X的第一个主成分为,Y的第一个主成分为![]() ,两者都经过了单位化。(这里的主成分并不是通过PCA得出的主成分)
,两者都经过了单位化。(这里的主成分并不是通过PCA得出的主成分)
3) ![]() ,
,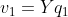 ,这一步看起来和CCA是一样的,但是这里的p和q都有主成分的性质,因此有下面4)和5)的期望条件。
,这一步看起来和CCA是一样的,但是这里的p和q都有主成分的性质,因此有下面4)和5)的期望条件。
4) ![]() ,即在主成分上的投影,我们期望是方差最大化。
,即在主成分上的投影,我们期望是方差最大化。
5) ![]() ,这个跟CCA的思路一致。
,这个跟CCA的思路一致。
6) 综合4)和5),得到优化目标 ![]() ,即协方差最大化。
,即协方差最大化。
形式化一点:

看起来比CCA还要简单一些,其实不然,CCA做完一次优化问题就完了。但这里的![]() 和
和![]() 对PLSR来说只是一个主成分,还有其他成分呢,那些信息也要计算的。
对PLSR来说只是一个主成分,还有其他成分呢,那些信息也要计算的。
先看该优化问题的求解吧:引入拉格朗日乘子

因此就是对称阵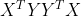 的最大特征值对应的单位特征向量,
的最大特征值对应的单位特征向量,![]() 就是
就是![]() 最大特征值对应的单位特征向量。
最大特征值对应的单位特征向量。
可见和![]() 是投影方差最大和两者相关性最大上的权衡,而CCA只是相关性上最大化。
是投影方差最大和两者相关性最大上的权衡,而CCA只是相关性上最大化。
求得了和![]() ,即可得到
,即可得到
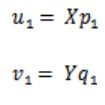
这里得到的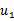 和
和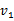 类似于上图中的绿色点,只是在绿色点上找到了X和Y的关系。如果就此结束,会出现与CCA一样的不能由X到Y映射的问题。
类似于上图中的绿色点,只是在绿色点上找到了X和Y的关系。如果就此结束,会出现与CCA一样的不能由X到Y映射的问题。
利用我们在PCA Revisited里面的第二种表达形式,我们可以继续做下去,建立回归方程:
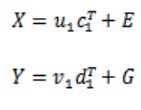
这里的c和d不同于p和q,但是它们之间有一定联系,待会证明。E和G是残差矩阵。
我们进行PLSR的下面几个步骤:




由上述步骤可知,虽然PLS多次迭代映射分解,但是最终组合起来,仍然是一个Y与X的线性回归Y=XB+F!将每次迭代过程中的映射变换的参数进行线性运算可以得到最终模型的参数B和F。
上述步骤中,矩阵C即为载荷矩阵(即通过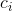 可计算出每个样本在第 i 个成分的得分)。
可计算出每个样本在第 i 个成分的得分)。
PLS结果的解读
(本部分查阅资料后总结)
通过PLS分析,可以得到PLS-VIP值和PLS-BETA值,可用于筛选变量(PLS-VIP最常使用)。
PLS-VIP
PLS-VIP指的是Variable Importance in Projection,最先在 3D QSAR in Drug Design: Theory Methods and Applications 这本书中提出(可惜这本书很贵,看不到相应的描述)。第 j 个变量的VIP值计算方法如下:
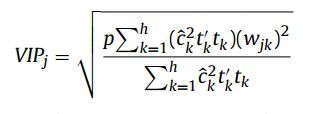
其中,p表示最初总共有p个变量参与分析;h表示最终总共进行了h次迭代计算(总共获得了h个维度);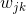 表示第k次迭代时(第k个维度),变量 j 进行映射时所采用的权重(即协方差矩阵中的系数),反应的是变量 j 对第k次映射结果
表示第k次迭代时(第k个维度),变量 j 进行映射时所采用的权重(即协方差矩阵中的系数),反应的是变量 j 对第k次映射结果 的解释程度;
的解释程度;![]() 表示第k次映射的结果 对
表示第k次映射的结果 对 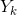 的解释程度( 注:是自变量X第k次的映射结果,是因变量Y在第k次迭代时被解释的部分 )。
的解释程度( 注:是自变量X第k次的映射结果,是因变量Y在第k次迭代时被解释的部分 )。
由于所有变量的VIP值的平方和等于1,所以往往将1作为VIP判定的界值:一般认为VIP小于1的变量对模型解释程度很低。
PLS-BETA
PLS-BETA值的是PLS最终回归模型中各个变量的系数。上一部分的推导结果可知,本质上最终的模型仍然是Y与X的线性回归Y=XB+F,这里的B即为系数向量。线性回归模型的系数越大往往预示着这个变量越重要(前提是各个变量已经标准化处理)。PLS-BETA 在许多文献里写作 Cov(t, Xi),即得分矩阵与自变量矩阵的的协方差。
Correlation Coefficient
Correlation Coefficient 指的是样品得分与变量之间的相关性(即预测的Y与每个变量 之间的相关性r),简称Corr.Coeffs,这其实是对PLS-BETA的补充。相关性越好,则说明该变量越重要。这个相关性还可以计算显著性p值(同理,PLS-BETA也可以计算p值),相关性r的界值没有规定,一般根据其对应的p值取0.05作为界值。
之间的相关性r),简称Corr.Coeffs,这其实是对PLS-BETA的补充。相关性越好,则说明该变量越重要。这个相关性还可以计算显著性p值(同理,PLS-BETA也可以计算p值),相关性r的界值没有规定,一般根据其对应的p值取0.05作为界值。
Corr.Coeffs在许多文献里也写作 Corr(t, Xi),即得分矩阵与自变量矩阵的的相关系数。根据协方差与相关系数的数学关系可知:Corr(t, Xi) = Cov(t, Xi) / (St * Sxi)。
V-Plot
同时参照VIP和Corr.Coeffs筛选变量,是常用的策略。由于大多数情况下,VIP值和Corr.Coeffs值相关性较好,且VIP恒正而Corr.Coeffs可正可负,因此可视化的图类似于V形,被称为V-Plot。
S-Plot
同时参照PLS-BETA和Corr.Coeffs筛选变量,也是常用的策略。由于PLS-BETA与Corr.Coeffs同正同负,因此可视化的图中所有的点全部分布在第一和第三象限,类似于S形,被称为S-Plot。
S-Plot 详细的解读,可以参考原始论文:Visualization of GC/TOF-MS-Based Metabolomics Data for Identification of Biochemically Interesting Compounds Using OPLS Class Models

原始论文部分截图如下:

Loading Plot 与 Score Plot
先回顾下PCA的 loading plot (可以参考:PCA - Loadings and Scores)。所谓 loading(载荷),指的是坐标映射的系数(权重),也就是原始变量与主成分之间的变换系数。对于每个主成分,都有一个载荷向量 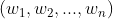 ,其中
,其中  表示 原始变量 的单位长度映射到该主成分坐标上所得值。若选择前两个主成分的载荷向量(为2*n的载荷矩阵),则可以绘制二维的 loading plot,其含义就是前两个主成分构成的二维坐标空间与原始变量构成的高维坐标空间之间的对应关系。可以看出,loading plot 是针对原始变量而言。
表示 原始变量 的单位长度映射到该主成分坐标上所得值。若选择前两个主成分的载荷向量(为2*n的载荷矩阵),则可以绘制二维的 loading plot,其含义就是前两个主成分构成的二维坐标空间与原始变量构成的高维坐标空间之间的对应关系。可以看出,loading plot 是针对原始变量而言。
PCA的二维 score plot 的含义是 以这两个主成分作为自变量绘制二维坐标系,各个样本的坐标值是多少。可以看出 score plot是针对样本而言。
常常将 loading plot 和 score plot 叠加在同一张图里,此时称为 bi-plot (如下图,图片出处不详)。

PLS 分析中,loading plot 和 score plot 的含义与PCA中一致,只是需注意 loading 不仅有 X loading,还有 Y loading。一般用w表示 X loading,用c表示 Y loading (比如 k分类时若选取2个主成分,则为2*k的矩阵),将二者叠加在同一张图(叠加不涉及任何计算,就是简单叠加),此时的 loading plot 又称为 w*c 图(参考自SIMCA-P中对w*c 图的解释)。另外,PLS的 score plot 中的坐标轴常用 t 表示,而不是 PC表示,但表达的是同一个意思,只是因为,在模型推导时,我们常常使用 t 矩阵表示 X 映射后的各个主成分。

图片来源:Plasma Amino Acid Profile in Patients with Aortic Dissection
这部分理解还可以参考:
成分分析中biplot函数
样本得分图和自变量的载荷图如何出现在一张图上
PLS模型评价
(本部分查阅资料后总结)
拟合效果
评价PLS-DA 模型拟合效果使用R2X、R2Y和Q2Y这三个指标,这些指标越接近1 表示PLS-DA 模型拟合数据效果越好。其中,R2X 和R2Y 分别表示PLSDA分类模型所能够解释X 和Y 矩阵信息的百分比,Q2Y 则为通过交叉验证计算得出,用以评价PLS-DA模型的预测能力,Q2Y 越大代表模型预测效果较好。
Q2和R2从计算上来看其实一模一样,只是采用的样本不一样,公式都是 1 - RSS/TSS(Q2中一般把RSS改写为PRESS,即 predicted residual error sum of squares)。R2是对 参与拟合的样本 计算残差,因此体现的是“拟合能力”;Q2是对 交叉验证中未参与拟合的样本 计算残差,因此体现的是“预测能力”。由于R2进行计算时,拟合后残差肯定不会变大,所以R2的取值在0-1之间;Q2中残差可能会比初始状态的残差更大,因此Q2甚至有可能为负数。另外,对于每个自变量都可以计算对应的R2和Q2。
从《Multi- and Megavariate Data Analysis Basic Principles and Applications》书中截取的公式如下(这本书比较权威):

完整的原文如下:

随着加入模型的变量增加(模型变复杂),R2会增加或进入平台期,但Q2可能会下降(Q2下降说明过拟合了),回想一下测试集和训练集的学习曲线可知。我们需要找一个平衡,其实也就是“方差variance”和“偏差bias”的平衡。

PCA分析中R2X >0.4为好(PCA中只有R2X,即最终纳入的几个成分累计对原始变量的还原程度 [或者说解释程度] );PLS-DA 和 OPLS-DA分析中,R2X 这个参数不重要了,主要是R2Y 和Q2,这两个值>0.5 为好,越接近1越好。我们经常看到PLS-DA中有个Q2(cum),是指建模后模型的预测能力(即最终得到线性模型Y=XB+F的预测能力),以大于0.5为宜,越接近1越好,cum 表示累积的意思,在公式中涉及到单自变量Q2的累乘运算,但其实就是模型整体的Q2。
是否过拟合
可靠的解读,需要进行permutation test(置换检验)判断模型是否过拟合。一般需检验模型的Q2值和R2值。对于Q2,要求置换检验结果的在y轴上的截距小于0,方可认为模型没有过拟合。这种标准相对严格,可能是考虑到:置换检验中标签打乱的比例和拟合/预测能力成正比例函数时(过原点坐标),过拟合情况正好消失(仅保留了该有的拟合能力),若稍微存在一定过拟合,则截距会在原点之上。但是,也有另一种宽松的标准,只要能推测出“只有极少数(5%)置换模拟的Q2值比当前Q2值大”,则认为没有过拟合(ropls包中采用的是这种宽松的方式,给出置换检验的p值进行判断)。
关于这个问题,笔者后来在《Multi- and Megavariate Data Analysis Basic Principles and Applications》书中得到了说法:根据经验来看,R2截距应小于0.3,Q2截距应小于0.05。


置换检验的基本原理:将真实分类结果(标签)屏蔽,重新随机赋予分类结果(标签),再进行建模。如果真实建模的Q2和随机标签建模的Q2接近,则说明模型过拟合。具体原理请参考其他资料。置换检验可视化的图,横坐标表示的是置换后的标签与真实标签的相似性(有多少比例的样本未打乱重新赋予标签)。

PLS-DA
PLS本质上是线性回归,若遇到分类任务,需要将因变量进行dummy处理(哑变量化),此时便称为PLS-DA。为了与PLS-DA区分,普通的PLS又称为PLSR。
笔者有个疑惑,对于二分类问题,直接使用PLSR进行处理是否合理?之前在SVM中也遇到过相似的疑惑:普通的二分类问题,用SVR处理是否合理?笔者之前的一些任务中,发现二分类中可以使用SVR替代SVM。但是解决这些疑惑还需要重新解读PLS-DA及SVR的原理,以及进行一些模拟数据的测试。
根据笔者的经验,将连续性因变量问题转为分类问题,可以仿照 Logistic,在原先回归的基础上再加一个连接函数(如Logistic 中的连接函数为logit 函数 [ 其反函数是 sigmoid 函数 ] ),将问题转为广义线性回归的任务。PLS 与 PLS-DA 之间的转换,为什么不采用这种技巧呢?
PLS 常用R工具包:ropls、pls、plsdepot 及 mixOmics;常用的python工具包:sklearn.cross_decomposition 中的 PLSRegression。
ropls包使用比较简单,可以跟着提供的案例进行学习。
pls 包是最基础的工具包,里面有封装了各种求解 PLS 的算法,其中的 CPPLS 算法可以解决多个因变量的问题及分类问题(分类问题常常使用哑变量法,被转换为多因变量问题)。但是 pls 包不能直接计算 vip 值,需要自己手动编写计算程序。
plsdepot 包也是值得推荐的工具包,里面同样封装了很多PLS 相关的算法。其中,plsreg1 可以处理普通的PLS任务,而plsreg2可以处理多个因变量的问题(也就可以处理分类问题)。另外,pslreg2 可以计算 vip 值,并且还计算了Q2等结果。
mixOmics 是另一个不错的工具,可以直接计算 PLS 和 PLS-DA等问题。mixOmics 中的vip方法可以计算 vip 值。
一个题外话——哑变量技巧
对于存在分类变量的问题,我们第一步就是对变量进行dummy处理。对于自变量的哑变量化与因变量的哑变量化,有一些细微的区别。
对于类别数为 k 的分类自变量,我们会设置(k-1)个哑变量:![]() 。哑变量的编码规则为:对于参照类别(又称类别0,可以人为定义一个类别作为计算的参照值,等价于连续性变量中的0值),我们将各个哑变量都赋值为0,即:
。哑变量的编码规则为:对于参照类别(又称类别0,可以人为定义一个类别作为计算的参照值,等价于连续性变量中的0值),我们将各个哑变量都赋值为0,即: ![]() ;对于其他类别,我们为每个类别选择一个哑变量,并将相应的哑变量赋值为1 而其他哑变量赋值 0,比如类别1可以这样赋值:
;对于其他类别,我们为每个类别选择一个哑变量,并将相应的哑变量赋值为1 而其他哑变量赋值 0,比如类别1可以这样赋值: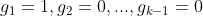 。这么处理是为了在结果解读时,能够为每个哑变量找到“参照值”。
。这么处理是为了在结果解读时,能够为每个哑变量找到“参照值”。
对于类别数为 k 的分类因变量,我们会设置 k 个哑变量: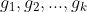 。为每个类别选择一个哑变量,并将相应的哑变量赋值为1 而其他哑变量赋值为 0。这种处理技巧很常见,理解也比较简单。比如 Logistic 的 softmax 函数也是采用哑变量的形式处理多分类问题。
。为每个类别选择一个哑变量,并将相应的哑变量赋值为1 而其他哑变量赋值为 0。这种处理技巧很常见,理解也比较简单。比如 Logistic 的 softmax 函数也是采用哑变量的形式处理多分类问题。
OPLS及OPLS-DA
(本部分出处忘记了)
OPLS-DA是PLS-DA的扩展,即首先使用正交信号校正技术,将X矩阵信息分解成与Y相关和不相关的两类信息,然后过滤掉与分类无关的信息,相关的信息主要集中在第一个预测成分。Trygg et al. (2002)认为该方法可以在不降低模型预测能力的前提下,有效减少模型的复杂性和增强模型的解释能力。与PLS-DA模型相同,可以用R2X、R2Y、Q2和OPLS-DA得分图来评价模型的分类效果。目前一般认为,在二分类问题中,OPLS-DA可以作为首选分析方法(代谢组学中更多人使用OPLS,而不是PLS)。
PLS的一些思考
PLS与LASSO的对比
(本部分原创)
PLS和LASSO,从本质上来讲,都是线性回归模型!只是说,这二者在进行回归建模时,顺便进行了特征降维。
在完成降维后,二者重新构建模型,得到的其实就是一个简单的线性回归模型。而在变量选择(降维)的过程中,两者采用的策略不同,PLS采用的是成分分解分析的思路,而LASSO采用的是惩罚约束策略。
至于具体哪种方法更好,很难说。在实际数据分析时,两种方法都可以进行尝试,最后选择效果更好的方法即可。
在应对共线性(collinearity)问题上,两者的表现如何呢?由于LASSO的原则是使模型尽可能简单,也就会尽可能消除共线性;但是PLS关注的是各个变量对Y的解释,会保留共线性的变量。2005年有篇论文(Performance of some variable selection methods when multicollinearity is present)对此进行了验证,结论里提到“The PLS-VIP method performed excellently in identifying relevant predictors and outperformed the other methods. It was also found that a model with good fitness performance may not guarantee good variable selection performance. Thus, for the purpose of selecting relevant process variables, process engineers must be careful when using model performance such as RMSE, R-squares, etc.” 该论文作者提出,在选择变量时,尽量避免使用R2等评价指标,而应该直接使用上述结果解读部分的方法。
PLS与GBDT的对比
理解了PLS的原理后,很容易发现PLS的思路很像集成学习Boosting,最典型比如GBDT(Gradient Boosting Decision Tree)。二者的思想都是先尽量拟合,然后一步步拟合残差部分,最后将各次的拟合集成。
朴素的思想:PLS和GBDT的拟合策略背后,都蕴含着一种由简入繁的思想,先拟合大致轮廓,再逐步拟合细节。
贪心的思想:局部最优化,PLS与GBDT都是考虑当前的最优拟合决策,这是典型的贪心拟合策略。贪心的解未必是全局最优解。
PLS与GBDT的不同:PLS本质上还是线性模型,GBDT可以是非线性的。PLS的生物学解释性较强,GBTD建立的模型未必能很好地解释(决策树类的模型关注决策条件的信息熵,往往会忽视变量的生物学解释;线性模型可以保留生物学线性关系,但无法考虑到潜在的交互作用 [除非添加交互项] )。
参考资料
偏最小二乘法回归(Partial Least Squares Regression)
N.L. Afanador, et al. Use of the bootstrap and permutation methods for a more robust variable importance in the projection metric for partial least squares regression. Analytica Chimica Acta. 2013
Il-Gyo Chong, et al. Performance of some variable selection methods when multicollinearity is present. Chemometrics and Intelligent Laboratory Systems. 2005
代谢组学数据分析的统计学方法综述
PCA - Loadings and Scores