OPLS-DA分析
美格基因·干货学习|一文读懂OPLS-DA分析 - 知乎 (zhihu.com)
官方教程 ropls: PCA, PLS(-DA) and OPLS(-DA) for multivariate analysis and feature selection of omics data (bioconductor.org)
一、ropls包简介
ropls包可以实现PCA、PLS(-DA)和OPLS(-DA)方法,包括R2和Q2质量指标,VIP值的计算,检测异常值的分数和正交距离,以及许多图形(分数、加载、预测、诊断、异常值等)。
二、背景知识
偏最小二乘法(PLS)是一种基于预测器和因变量之间协方差的潜在变量回归方法,已被证明可以有效地处理多共线预测器的数据集,如光谱测量。
正交偏最小二乘(OPLS)算法,以分别对与因变量相关和正交的预测器的变化进行建模。
OPLS与PLS具有相似的预测能力,并改进了对预测成分和系统变化的解释。特别是,单个因变量的OPLS建模只需要一个预测组件。PCA是一种无监督降维方法,对相关性较小的变量不敏感,而PLS-DA能够有效解决这个问题。而OPLS-DA结合了正交信号和PLS-DA来筛选差异变量,是一种有监督的判别分析统计方法。运用偏最小二乘回归建立代谢物表达量与样本类别之间的关系模型,实现对样本类别的预测。本分析主要用于代谢组学中差异代谢物的筛选。OPLS-DA需要样本变量矩阵和样本分类矩阵两个文件确立样本关系。
Q2Y指标和排列测试等诊断对于避免过度拟合和评估模型的统计意义非常重要。
投影中的可变重要性(VIP)反映了每个组件的加载权重以及该组件解释的响应的可变性,可用于特征选择。
三、分析实例
1-准备工作
液相色谱高分辨质谱法(LTQ Orbitrap)分析了来自183位成人的尿液样品。
sacurine list 包含了三个数据矩阵。
dataMatrix为样本-代谢物含量矩阵(log10转换过),记录了各种类型的代谢物在各样本中的含量信息。共计183个样本(行)以及109种代谢物(列)。
sampleMetadata中记录了183个样本所来源个体的年龄、体重、性别等信息。
variableMetadata为109种代谢物的注释详情,MSI level水平。
#加载包
rm(list = ls())
# load packages
library(ropls)
# load data
data(sacurine)
attach(sacurine)
#查看数据集
head(sacurine$dataMatrix[ ,1:2])
head(sacurine$sampleMetadata)
head(sacurine$variableMetadata)2-PCA
#PCA分析
sacurine.pca <- opls(dataMatrix)
#根据性别标记/着色,并绘制男性和女性亚组的椭圆图。
genderFc <- sampleMetadata[, "gender"]
plot(sacurine.pca,
typeVc = "x-score",
parAsColFcVn = genderFc)
#可调整绘图风格
plot(sacurine.pca,
typeVc = "x-score",
parAsColFcVn = genderFc,
parLabVc = as.character(sampleMetadata[, "age"]),
parPaletteVc = c("green4", "magenta"))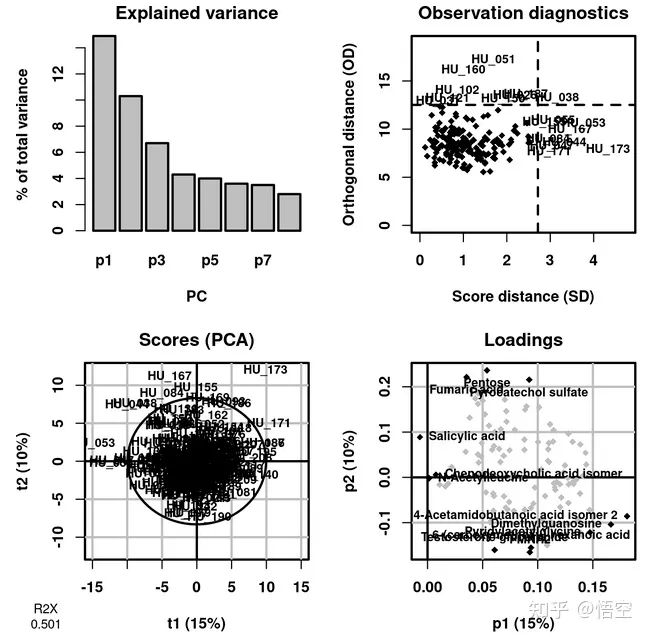
左上碎石图:说明前几个能解释大部分变量。
右上离群值:此图显示了距离并且与投影平面正交,样本的名称说明该样本至少其中一个距离超出正常范围。
左下x-score:沿每个轴的方差等于由每个成分表示的方差:因此它取决于矩阵X的总方差以及成分代表该方差的百分比; 当从一个成分到另一个更高指数的成分时,x-score会降低。
右下 x-loading:3个变量每次加载的最极端值(正值和负值)为黑色并带有标签。
注意
- 由于 dataMatrix 不包含缺失值,因此奇异值默认使用分解; NIPALS 可以用
algoC指定算法。 predI = NAPCA预测成分的默认数量意味着将计算成分(最多10个)直到累积方差超过 50%。 如果在第 10 个组件中没有达到 50%, 将发出警告消息(仍可以通过指定predI值计算以下成分)。

3-PLS和PLS-DA
sacurine.plsda <- opls(dataMatrix, genderFc)
左上inertia条形图:此处的图形表明 3 个正交分量可能足以代表大部分信息,展示累计解释率评估正交组分是否足够。
右上significance显著性诊断:模型的R2Y和Q2Y与y因变量随机排列后获取的对应值相比较,实际和模拟模型的R2Y和Q2Y值经随机排列后的散点图,模型R2Y和Q2Y(散点)大于真实值时(横线),表明产生过拟合。
左下 outlier诊断:高值的样本标注出名称。
右下 x-score图:成分数量和累积R2X、R2Y和**Q2Y,**各样本在OPLS-DA轴中的坐标,颜色代表性别分组。
注意
R2X和R2Y分别表示所建模型对X和Y矩阵的解释率,Q2表示模型的预测能力,它们的值越接近于1表明模型的拟合度越好,训练集的样本越能够被准确划分到其原始归属中。permI参数可调节随机排列次数。
4-OPLS和OPLS-DA
# 分组以性别为例
# 通过orthoI指定正交组分数目
# orthoI = NA时,执行OPLS,并通过交叉验证自动计算适合的正交组分数
oplsda = opls(dataMatrix, genderFc, predI = 1, orthoI = NA)
#评估模型的预测性能,首先训练在样本子集上建模
sacurine.oplsda <- opls(dataMatrix, genderFc,
predI = 1, orthoI = NA,
subset = "odd")
#检查训练子集的预测
trainVi <- getSubsetVi(sacurine.oplsda)
table(genderFc[trainVi], fitted(sacurine.oplsda))
#计算测试子集的性能
table(genderFc[-trainVi],
predict(sacurine.oplsda, dataMatrix[-trainVi, ]))
注意
- 对于单个y的OPLS建模,预测成分的数量为1。
- 预测分量显示为横坐标;正交分量作为纵坐标。
5-使用ggplot2进行可视化
library(ggplot2)
library(ggsci)
library(tidyverse)
#提取样本在 OPLS-DA 轴上的位置
sample.score = oplsda@scoreMN %>% #得分矩阵
as.data.frame() %>%
mutate(gender = sacurine[["sampleMetadata"]][["gender"]],
o1 = oplsda@orthoScoreMN[,1]) #正交矩阵
head(sample.score)#查看
p <- ggplot(sample.score, aes(p1, o1, color = gender)) +
geom_hline(yintercept = 0, linetype = 'dashed', size = 0.5) + #横向虚线
geom_vline(xintercept = 0, linetype = 'dashed', size = 0.5) +
geom_point() +
#geom_point(aes(-10,-10), color = 'white') +
labs(x = 'P1(5.0%)',y = 'to1') +
stat_ellipse(level = 0.95, linetype = 'solid',
size = 1, show.legend = FALSE) + #添加置信区间
scale_color_manual(values = c('#008000','#FFA74F')) +
theme_bw() +
theme(legend.position = c(0.1,0.85),
legend.title = element_blank(),
legend.text = element_text(color = 'black',size = 12, family = 'Arial', face = 'plain'),
panel.background = element_blank(),
panel.grid = element_blank(),
axis.text = element_text(color = 'black',size = 15, family = 'Arial', face = 'plain'),
axis.title = element_text(color = 'black',size = 15, family = 'Arial', face = 'plain'),
axis.ticks = element_line(color = 'black'))
p
6-VIP值筛选差异代谢物
#6-1-展示VIP值分布情况
ageVn <- sampleMetadata[, "age"]
pvaVn <- apply(dataMatrix, 2,
function(feaVn) cor.test(ageVn, feaVn)[["p.value"]])
vipVn <- getVipVn(opls(dataMatrix, ageVn,
predI = 1, orthoI = NA,
fig.pdfC = "none"))
quantVn <- qnorm(1 - pvaVn / 2)
rmsQuantN <- sqrt(mean(quantVn^2))
opar <- par(font = 2, font.axis = 2, font.lab = 2,
las = 1,
mar = c(5.1, 4.6, 4.1, 2.1),
lwd = 2, pch = 16)
plot(pvaVn, vipVn,
col = "red",
pch = 16,
xlab = "p-value", ylab = "VIP", xaxs = "i", yaxs = "i")
box(lwd = 2)
curve(qnorm(1 - x / 2) / rmsQuantN, 0, 1, add = TRUE, col = "red", lwd = 3)
abline(h = 1, col = "blue")
abline(v = 0.05, col = "blue")
par(opar)
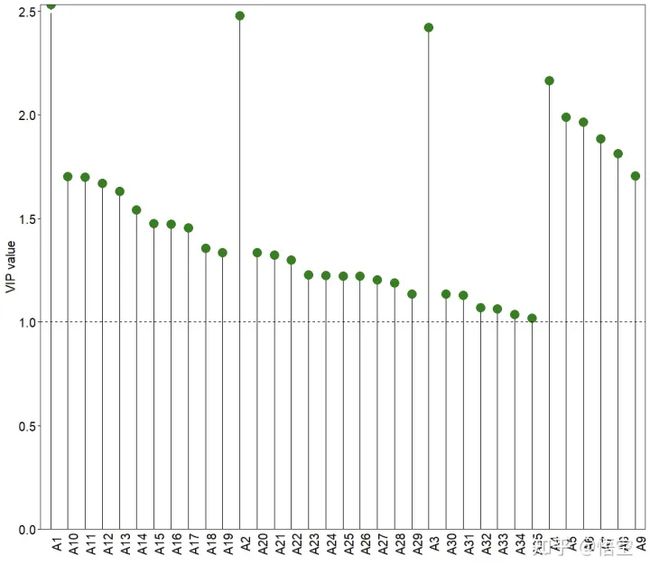
#6-2-绘制棒棒糖图VIP值筛选差异代谢物
#VIP值帮助寻找重要的代谢物
vip <- getVipVn(oplsda)
vip_select <- vip[vip > 1] #通常以VIP值>1作为筛选标准
head(vip_select)
vip_select <- cbind(sacurine$variableMetadata[names(vip_select), ], vip_select)
names(vip_select)[4] <- 'VIP'
vip_select <- vip_select[order(vip_select$VIP, decreasing = TRUE), ]
head(vip_select) #带注释的代谢物,VIP>1 筛选后,并按 VIP 降序排序
#对差异代谢物进行棒棒糖图可视化
#代谢物名字太长进行转换
vip_select$cat = paste('A',1:nrow(vip_select), sep = '')
p2 <- ggplot(vip_select, aes(cat, VIP)) +
geom_segment(aes(x = cat, xend = cat,
y = 0, yend = VIP)) +
geom_point(shape = 21, size = 5, color = '#008000' ,fill = '#008000') +
geom_point(aes(1,2.5), color = 'white') +
geom_hline(yintercept = 1, linetype = 'dashed') +
scale_y_continuous(expand = c(0,0)) +
labs(x = '', y = 'VIP value') +
theme_bw() +
theme(legend.position = 'none',
legend.text = element_text(color = 'black',size = 12, family = 'Arial', face = 'plain'),
panel.background = element_blank(),
panel.grid = element_blank(),
axis.text = element_text(color = 'black',size = 15, family = 'Arial', face = 'plain'),
axis.text.x = element_text(angle = 90),
axis.title = element_text(color = 'black',size = 15, family = 'Arial', face = 'plain'),
axis.ticks = element_line(color = 'black'),
axis.ticks.x = element_blank())
p2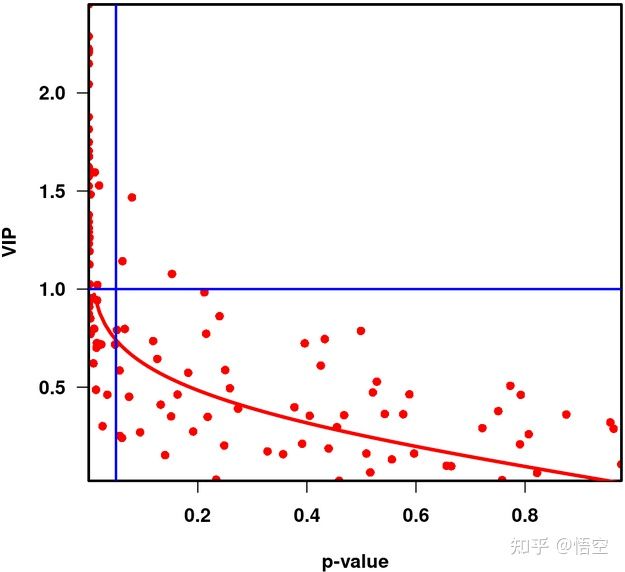
上图左上区域是关心的值,下图对这些点进行展示。
四、其他结果图信息
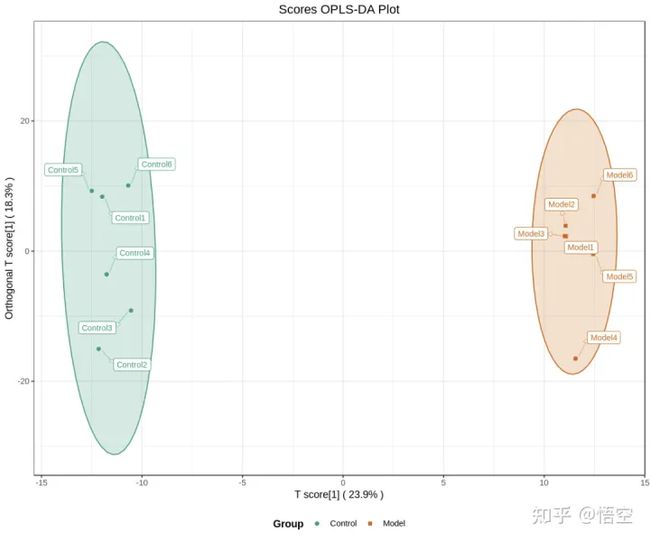
1-OPLS-DA得分图
横坐标反映组间差异;纵坐标反映组内差异。
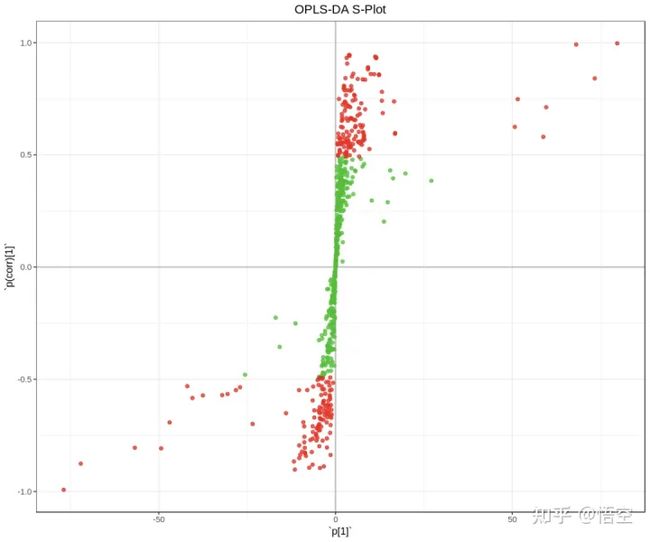
2-OPLS-DA的S-plot图
越靠近两个角的代谢物重要度越强。红色的点表明这些代谢物的VIP值大于等于1 , 绿色的点表示这些代谢物的VIP值小于等于1。
3-OPLS-DA的模型验证permutation Test图
模型验证 permutation Test 图的横坐标表示模型的准确率,纵坐标表示200次 permutation Test 中200个模型的准确率的频数,箭头表示本OPLS-DA模型准确率所在的位置。其中R2X和R2Y分别表示所建模型对X和Y矩阵的解释率, Q2表示模型的预测能力,理论上R2、Q2数值越接近1说明模型越好,越低说明模型的拟合准确性越差。通常情况下 , R2、Q2高于0.5较好,高于0.4即可接受。

参考内容来源
ropls: PCA, PLS(-DA) and OPLS(-DA) for multivariate analysis and feature selection of omics data
理解OPLS-DA分析的原理和结果图
木舟笔记:R实战 | OPLS-DA(正交偏最小二乘判别分析)筛选差异变量(VIP)及其可视化