Emvirus: 基于 embedding 的神经网络来预测 human-virus PPIs【Biosafety and Health,2023】

研究背景:
- Human-virus PPIs 预测对于理解病毒感染机制、病毒防控等十分重要;
- 大部分基于 machine-learning 预测 human-virus PPIs 的方法利用手动方法处理序列特征,包括统计学特征、系统发育图谱、理化性质等;
- 本文作者提出了一个名为
Emvirus的方法,它利用Doc2Vec获取蛋白序列特征,并将序列特征输入到由 CNN 和 Bi-LSTM 构成的网络中预测 human-virus PPIs;
数据集构成:
- 正样本 human-virus PPIs 来自 Yang et.al 收集的多个来源(包括 PHISTO,VirHostNet,VirusMentha,HPIDB,PDB以及一些实验数据)的 PPIs,去掉重复的和无统计学显著性的 PPIs 之后,最终得到 27493 对正样本 PPIs。
- 负样本 human-virus PPIs 来自 Yang et.al 中使用的基于 dissimilarity‐based negative sampling method 构建的负样本 PPIs。
- 正样本:负样本 = 1:10
- 训练集:测试集 = 20:1
- 对于数据集中样本类别不平衡的处理办法:作者利用 SMOTE 方法对正样本进行过采样,构建 balanced training datasets。

研究思路和方法:

论文代码:https://github.com/hongjiala/PPIs
1. 利用 Doc2vec 获取蛋白质序列的特征向量:
Doc2vec 是NLP中Word2vec方法的拓展,相比于 Word2vec,Doc2vec 可以从不同长度的蛋白序列中学到固定长度的序列特征表示。
(至于图中蛋白向量形状为 1x3000,暂时没想清楚怎么来的)
#【本段代码来自 作者提供的 doc2vec/doc2vec.py,我只是添加了一些注释信息。】
# -*- coding: utf-8 -*-
"""
Created on Tue May 26 18:19:53 2022
@author: xiepengfei
"""
import numpy as np
from Bio import SeqIO
from nltk import trigrams, bigrams,ngrams ## 用来给氨基酸序列进行分词
from keras.preprocessing.text import Tokenizer
from gensim.models import Word2Vec
import re
from gensim.models.doc2vec import Doc2Vec, TaggedDocument ## 用于 embedding
from gensim.test.utils import get_tmpfile
np.set_printoptions(threshold=np.inf)
## 将 每条氨基酸序列 划分成小片段,之间以空格分开,并将每一个病毒中的所有的序列保存在 texts 列表中
names = ["DENV","Hepatitis","Herpes","HIV","Influenza","Papilloma","SARS2","ZIKV"] ## 有这些病毒的序列,每个病毒序列都单独处理,训练embdding模型
for name in names:
texts = []
for index, record in enumerate(SeqIO.parse('fasta/%s.fasta'%name, 'fasta')):
tri_tokens = ngrams(record.seq,6) ## 将蛋白质序列连续分割成长度为6的片段
temp_str = ""
for item in ((tri_tokens)): ## item 就是每一条氨基酸序列的片段,格式 ("A","B","C","D","E","F"), ("B","C","D","E","F","G"), ("C","D","E","F","G","H")
# print(item),
items = "" ## items 就是将每个片段中的氨基酸残基字符拼接成一个字符串,即 "ABCDEF", "BCDEFG", "CDEFGH"
for strs in item:
items = items+strs
temp_str = temp_str + " " + items ## 将氨基酸片段字符串拼起来,之间以空格分开,即 "ABCDEF BCDEFG CDEFGH"
#temp_str = temp_str + " " +item[0]
texts.append(temp_str) ## 将 temp_str 保存到 texts 中,格式:["ABCDEF BCDEFG CDEFGH", "ABCDEF BCDEFG"]
## 将 texts 中保存的氨基酸序列中的一些特殊字符(stop中列举的特殊字符)去掉,结果保存在 seq 中 (seq中的内容和texts中的内容的区别就是前者没有那些特殊字符)
seq=[]
stop = '[’!"#$%&\'()*+,-./:;<=>?@[\\]^_`{|}~]+'
for doc in texts:
doc = re.sub(stop, '', doc)
seq.append(doc.split())
documents = [TaggedDocument(doc, [i]) for i, doc in enumerate(seq)] ## 将 seq 列表中的每条序列转化为 TaggedDocument对象,words就是每条序列doc,tags就是序列的索引[i]
model = Doc2Vec(documents , vector_size=1000, window=500, min_count=1, workers=12) ## 构建 Doc2vec 模型
model.train(documents ,total_examples=model.corpus_count, epochs=50) ## 训练模型
#model.save("autodl-tmp/my_doc2vec_model.model") # you can continue training with the loaded model!
#model.dv.save_word2vec_format('%s.vector'%name)
# test_seq = ['MRQGCKFRGSSQKIRWSRSPPSSLLHTLRPRLLSAEITLQTNLPLQSPCCRLCFLRGTQAKTLK']
# # test_text = ngrams(test_seq,6)
# # temp_str_test = ""
# # for item in ((test_text)):
# # # print(item),
# # print(item)
# # items = ""
# # for strs in item:
# # items = items+strs
# # temp_str = temp_str_test + " " + items
# inferred_vector_dm = model.infer_vector(test_seq)
# print(inferred_vector_dm)
np.save("vec/new_%s_vector.npy"%name,model.dv.vectors) ## 保存特征向量
2. 将 human-virus PPI pairs 转化为 feature vector pairs
将 human 的蛋白向量、virus 的蛋白向量、标签放到一起。
如下代码所示:(详情见:https://github.com/hongjiala/PPIs/blob/master/pair/form_pair_data.py)

3. 用 SMOTE 方法对正样本进行过采样:
这部分代码是用MATLAB写的,看不太懂。详情见:https://github.com/hongjiala/PPIs/tree/master/smote
关于SMOTE的原理(参考:arXiv:1106.1813):
This paper shows that a combination of our method of over-sampling the minority (abnormal) class and under-sampling the majority (normal) class can achieve better classifier performance (in ROC space) than only under-sampling the majority class.
This approach is inspired by a technique that proved successful in handwritten character recognition (Ha & Bunke, 1997). They created extra training data by performing certain operations on real data. In their case, operations like rotation and skew were natural ways to perturb the training data.
We generate synthetic examples in a less application-specific manner, by operating in “feature space” rather than “data space”.
The minority class is over-sampled by taking each minority class sample and introducing synthetic examples along the line segments joining any/all of the k minority class nearest neighbors. Depending upon the amount of over-sampling required, neighbors from the k nearest neighbors are randomly chosen.

简单来说的话就是:在原始样本的 “feature space” 中某个样本点 i i i的最近邻的 k k k个样本点中随机的一个点 n n nn nn,计算 n n nn nn和 i i i在 “feature space” 中的特征差值 d i f dif dif,然后生成0-1之间随机数 g a p gap gap,则新生成的样本点 n e w i n d e x newindex newindex的特征值 = i i i的特征值 + g a p gap gap * d i f dif dif
4. 构建模型:
由 CNN、Attention 和 Bi-LSTM 构建模型,详情见(https://github.com/hongjiala/PPIs/blob/master/train/model_protein.py)

5. 训练并测试模型:
详情见:https://github.com/hongjiala/PPIs/tree/master/train
实验结果及讨论:
作者对每一种病毒都用相同的神经网络框架分别训练了一个模型,每个病毒对应的模型的预测结果:
1. 各模型对各自的 human-virus PPIs 预测结果:
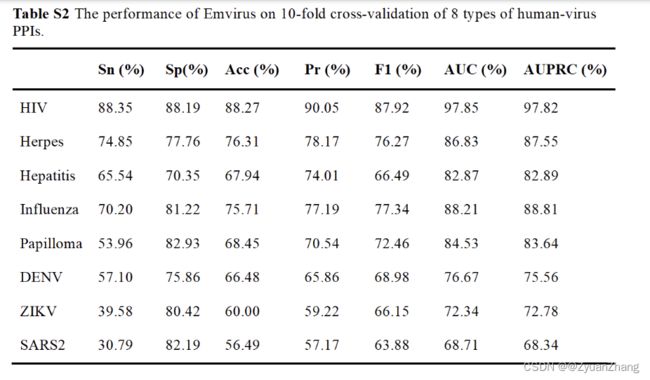
2. 不同特征及不同模型对各病毒的 human-virus PPIs 预测结果:

3. 各模型跨病毒的 human-virus PPIs 预测结果:

讨论:
- 基于 Doc2Vec + CNN + Bi-LSTM 的方法构建的模型,并以不同病毒的 human-virus PPIs 数据分别对模型进行训练,除了某些病毒以外,大部分的病毒的模型的预测效果挺好的。
- 与手动抽取序列特征的方法(如 PSSM,LD,CT,AC等)相比,用 Doc2Vec 可以更好地获取序列特征。
- 用不同病毒的 PPIs 数据训练的模型在进行跨病毒的 human-virus PPIs 预测的时候,模型基本没有分辨能力,即模型的泛化能力较差,可能是由于不同病毒的 human-virus PPIs 的数据分布或者特征组成差别较大导致的。
- 整体上而言,Doc2Vec + LSTM 可以对某些特定的病毒实现比较好的 human-virus PPIs 预测。