8+孟德尔随机化+转录组,双样本和多元MR分析。
今天给同学们分享一篇孟德尔随机化+转录组的生信文章“Exploring the causality and pathogenesis of systemic lupus erythematosus in breast cancer based on Mendelian randomization and transcriptome data analyses”,这篇文章于2023年1月16日发表在Front Immunol期刊上,影响因子为8.786。

乳腺癌是全球女性最常见的癌症,与慢性炎症密切相关。由于女性中SLE的高发病率,积累的观察性或队列研究已经探讨了SLE和乳腺癌之间的关联。然而,观察性研究得出了相互矛盾的结论,因为结果可能受到许多潜在混杂因素的影响,包括样本大小和抗SLE免疫抑制治疗。SLE和乳腺癌的流行病学模式在不同种族人群之间也可能有所不同。因此,需要采用更加精心设计的方法来评估系统性红斑狼疮与乳腺癌风险之间的因果关系。
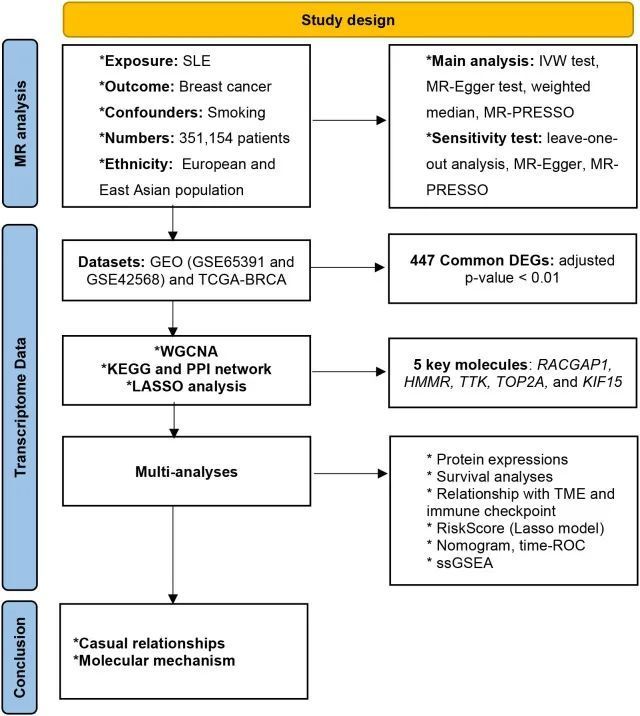
图1 研究设计概述
1. SNP的选择
总的来说,这项研究分析了共计243,218名欧洲血统个体(128,178例患者和115,040例对照组)以及107,936名东亚血统个体(9,774例患者和98,162例对照组)。作者从全基因组关联研究中提取了与SLE显著相关的IVs(p< 5 × 10 −8 ),并去除了连锁不平衡(r 2 <0.001,10,000-kb)。此外,作者的分析中F统计量大于100,表明这些IVs能够强有力地预测SLE的发病率。
2. 遗传易感性与SLE和乳腺癌风险
MR分析显示欧洲队列中SLE与乳腺癌之间不存在因果关联(乳腺癌:OR 0.9985,95%CI 0.9873-1.0099,p=0.79;ER+乳腺癌:OR 0.9974,95%CI 0.9850-1.0101,p=0.69;ER-乳腺癌:OR 1.009,95%CI 0.99-1.02,p=0.22)。没有证据表明其他MR方法基于乳腺癌风险的增加。然而,注意到东亚人群中SLE和乳腺癌的遗传易感性的因果推断(IVW:OR:0.95,95%CI:0.92-0.98,p=0.006;加权中位数:OR:0.93,95%CI:0.88-0.97,p=0.002;MR-PRESSO:OR 0.95,95%CI:0.92-0.98,p=0.004)(图2)。多变量MR分析也支持了SLE与东亚人群中乳腺癌的显著关联发现(SNPs:25,OR:0.95,95%CI:0.92-0.98,p=0.0013),在调整混杂因素(吸烟,特征ID:ieu-b-4877)后。

图2 系统性红斑狼疮对东亚人乳腺癌风险的因果关系
3. MR估计的敏感性分析
首先,作者进行了MR-Egger回归分析,以研究水平多效性,并且结果证实多效性不太可能对因果关系产生偏倚(所有p值>0.05)。其次,MR-PRESSO测试的结果与无异常值的IVW方法一致,表明原始结果可靠。第三,考虑到东亚队列中SLE和乳腺癌之间的潜在关系,作者进行了逐个排除分析和Cochrane Q检验。逐个排除分析发现没有单个SNP驱动SLE和乳腺癌之间的因果关系(图2)。Cochrane Q检验的p值均大于0.05(IVW测试的Q值:32.93,p=0.2;MR-Egger测试的Q值:32.8,p=0.17),表明SNP之间没有异质性。
4. 乳腺癌患者中SLE相关DEGs的鉴定
在标准化微阵列结果后(图3A、B),识别出SLE和乳腺癌相关数据集之间的447个常见差异表达基因(图3C)。通过聚类,WGCNA(软阈值功率=6)进一步去除了灰色模块中的61个明显异常值,并识别出386个感兴趣的中心基因(图3D、E)。
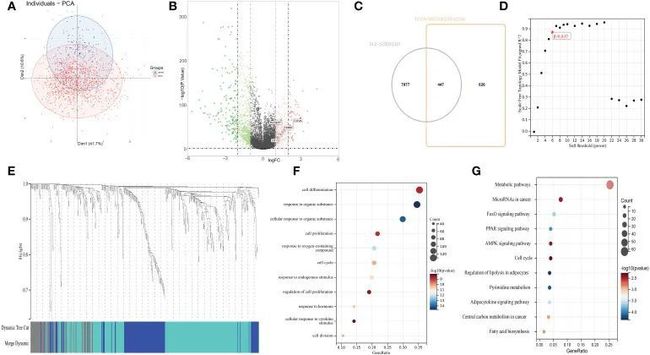
图3
5. 功能特点分析
为了进一步了解乳腺癌中386个SLE-DEGs的潜在功能,作者进行了GO和KEGG富集分析。GO分析显示DEGs富集在细胞周期、细胞增殖和对激素的反应方面(图3F)。KEGG富集分析主要涉及与癌症和细胞周期相关的途径,包括代谢途径、微小RNA、转录调控失调、蛋白聚糖和中心碳代谢(图3G)。
6. PPI网络和关键基因分析
首先,对386个常见的差异表达基因构建了PPI网络。其次,使用Cytoscape的Cytuhubba插件计算了前20个中心基因(AURKA、UBE2C、CDC20、PTTG1、CCNB2、MELK、NDC80、CENPF、PRC1、KIF23、TOP2A、RACGAP1、NUSAP1、HMMR、ASPM、KIF15、TTK、DLGAP5、CCNA2和NCAPG)(图4A)。第三,斯皮尔曼相关分析显示这二十个中心基因之间存在显著密切的关联(所有p值<0.0001)(图4B)。

图4
7. 预测模型的构建和验证
LASSO回归方法被用来优化20个中心基因。最终,选择了最有价值的五个预测基因(RACGAP1、HMMR、TTK、TOP2A和KIF15)来构建SLEscore(图4C、D)。SLEscore的五个中心基因在肿瘤样本中均显著上调(图4E-G)。根据SLEscore,乳腺癌患者被分为两个亚组,其中SLEscore high 与五个预后分子的高表达水平相关(图5A)。与低SLEscore组相比,高SLEscore组与明显更差的总生存期相关(图5B)。ROC曲线表明,SLEscore可以作为预测乳腺癌患者总生存期的敏感标志物(3年AUC:0.81,5年AUC:0.91)(图5C)。此外,多变量COX回归分析表明,SLEscore是乳腺癌患者的独立风险因素(HR 7.1,95%CI 1.50-33.4,p=0.013)(图5D)。作者建立的SLEscore的C指数为0.73(标准误差:0.043)。在GSE42568数据集中进一步验证了SLEscore,表明使用TCGA数据库构建的SLEscore是乳腺癌患者的独立预后因子(HR 1.92,95%CI 1.08-3.42,p=0.02)(图5E)。随后,作者通过整合SLEscore、年龄和TNM分期为乳腺癌患者建立了一个预测模型,该模型在预测乳腺癌患者1年、3年和5年的生存率方面表现良好(图6A、B)。
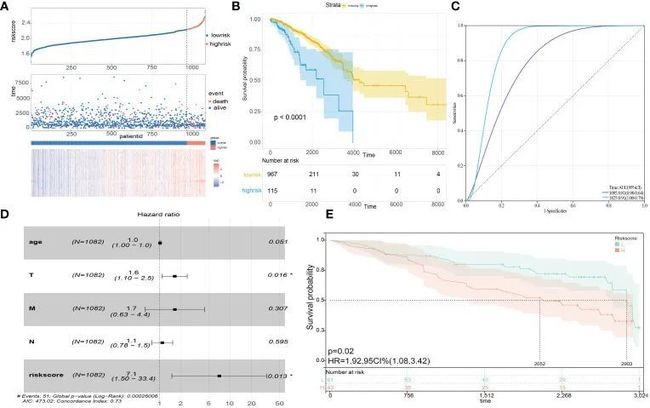
图5
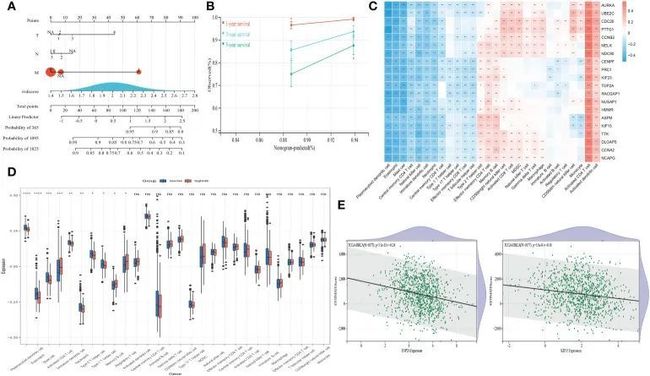
图6
8. TME细胞的探索,ESTIMATE评分,ICB和PANoptosis
Spearman相关分析揭示了20种分子与TME浸润细胞之间的显著关联(图6C)。值得注意的是,高SLE得分与较低的树突状细胞、嗜酸性粒细胞、肥大细胞、CD4+ T细胞和辅助T细胞表达显著相关(图6D)。ESTIMATE得分与五个中心基因的表达水平呈负相关(所有p值<0.05)(图6E),表明其与疾病结果和肿瘤浸润免疫环境的关系。SLE得分与8种ICB和PANoptosis基因模式的相关性显著,这些已被证明是乳腺癌患者预后生物标志物。
总结
作者的MR分析表明,SLE患者在东亚人群中对乳腺癌的风险较低。本研究还为乳腺癌和SLE患者的分层提供了路线图,有助于改进个体化随访和个性化决策的策略。