0046-【宏基因组】-qiime2官方教程实践1-Moving pictures of the human microbiome
1. 数据文章——Moving pictures of the human microbiome
文章下载:https://www.ncbi.nlm.nih.gov/pubmed/21624126
样本选取:
2个生物个体,4个部分,396个时间点
文库构建:
Meta16S V4区文库
测序策略:
illunima 、454
不同区域,选择不同的二代测序平台
信息分析:
qiime2-2018.4 软件,及测试代码集
注:与中文的帮助文档2017.7代码有少量不同
本次测试:
本示例的的数据来自文章《Moving pictures of the human microbiome》,Genome Biology 2011,取样来自两个人身体四个部位五个时间点。
2. 流程实践
1.准备数据
# 下载实验设计表
wget http://bailab.genetics.ac.cn/markdown/sample-metadata.tsv
# 下载实验测序数据
mkdir -p emp-single-end-sequences
wget -O "emp-single-end-sequences/barcodes.fastq.gz" "https://data.qiime2.org/2017.7/tutorials/moving-pictures/emp-single-end-sequences/barcodes.fastq.gz"
wget -O "emp-single-end-sequences/sequences.fastq.gz" "https://data.qiime2.org/2017.7/tutorials/moving-pictures/emp-single-end-sequences/sequences.fastq.gz"
# 生成qiime需要的artifact文件(qiime文件格式,将原始数据格式标准化)
qiime tools import \
--type EMPSingleEndSequences \
--input-path emp-single-end-sequences \
--output-path emp-single-end-sequences.qza输出显示:
$head sample-metadata.tsv
#SampleID BarcodeSequence LinkerPrimerSequence BodySite Year Month Day Subject ReportedAntibioticUsage DaysSinceExperimentStarDescription
L1S8 AGCTGACTAGTC GTGCCAGCMGCCGCGGTAA gut 2008 10 28 subject-1 Yes 0 subject-1.gut.2008-10-28
L1S57 ACACACTATGGC GTGCCAGCMGCCGCGGTAA gut 2009 1 20 subject-1 No 84 subject-1.gut.2009-1-20
L1S76 ACTACGTGTGGT GTGCCAGCMGCCGCGGTAA gut 2009 2 17 subject-1 No 112 subject-1.gut.2009-2-17
total 28M
-rw-rw-r-- 1 toucan toucan 3.7M Jul 22 2017 barcodes.fastq.gz
-rw-rw-r-- 1 toucan toucan 25M Jul 22 2017 sequences.fastq.gz
# fastq序列文件
1 @HWI-EAS440_0386:1:23:17547:1423#0/1
2 TACGNAGGATCCGAGCGTTATCCGGATTTATTGGGTTTAAAGGGAGCGTAGATGGATGTTTAAGTCAGTTGTGAAAGTTTGCGGCTCAACCGTAAAATTGCAGTTGATACTGGATATCTTGAGTGCAGTTGAGGCAGGGGGGGA
3 +
4 IIIE)EEEEEEEEGFIIGIIIHIHHGIIIGIIHHHGIIHGHEGDGIFIGEHGIHHGHHGHHGGHEEGHEGGEHEBBHBBEEDCEDDD>B?BE@@B>@@@@@CB@ABA@@?@@=>?08;3=;==8:5;@6?##############
5 @HWI-EAS440_0386:1:23:14818:1533#0/1
6 CCCCNCAGCGGCAAAAATTAAAATTTTTACCGCTTCGGCGTTATAGCCTCACACTCAATCTTTTATCACGAAGTCATGATTGAATCGCGAGTGGTCGGCAGATTGCGATAAACGGGCACATTAAATTTAAACTGATGATTCCAC
7 +
8 64<2$24;1)/:*BBDD##################################################################################################################### 数据标准化为qiime2的输入数据
# EMPSingleEndSequences——单端测序、EMPPairedEndSequences——双端测序
# 输入文件夹路径、输出文件
# qza为二进制文件,不能直接打开
qiime tools import --type EMPSingleEndSequences --input-path emp-single-end-sequences --output-path emp-single-end-sequences.qza可视化qza文件网站:https://view.qiime2.org/
可视化后显示,
- Peek 记录了 文件的类型
- 流程图
name:"emp-single-end-sequences.qza"
uuid:"207517a2-5d10-43dc-93c3-74a176fcfb6c"
type:"EMPSingleEndSequences"
format:"EMPSingleEndDirFmt"2. 拆分样品
# 按barcode拆分样品 Demultiplexing sequences
qiime demux emp-single \
--i-seqs emp-single-end-sequences.qza \ --m-barcodes-file sample-metadata.tsv \ --m-barcodes-category BarcodeSequence \ --o-per-sample-sequences demux.qza
# 结果统计
qiime demux summarize \
--i-data demux.qza \ --o-visualization demux.qzv
# 查看结果 (依赖XShell+XManager或其它ssh终端和图形界面软件)
qiime tools view demux.qzv结果显示:

3. 序列质控和生成OTU表
# 单端序列去噪, 去除左端0bp(--p-trim-left用于切除边缘低质量区),序列切成120bp长;生成代表序列和OTU表;并重命名用于下游分析
# denoise-single——单端模式
# --i-demultiplexed-seqs 输入序列
# --p-trim-left 左边切除长度为0,等于不切除
# --p-trunc-len 长度过滤最小值
# --o-representative-sequences 代表序列输出文件路径
# --o-table 特征标文件路径
# -o-denoising-stats 噪声统计
qiime dada2 denoise-single \
--i-demultiplexed-seqs demux.qza \
--p-trim-left 0 \
--p-trunc-len 120 \
--o-representative-sequences rep-seqs-dada2.qza \
--o-table table-dada2.qza \
--o-denoising-stats stats-dada2.qza
mv rep-seqs-dada2.qza rep-seqs.qza
mv table-dada2.qza table.qza统计文件可视化
qiime metadata tabulate \
--m-input-file stats-dada2.qza \
--o-visualization stats-dada2.qzv统一命名
mv rep-seqs-dada2.qza rep-seqs.qza
mv table-dada2.qza table.qza4. Feature表统计、代表序列统计
qiime feature-table summarize \
--i-table table.qza \
--o-visualization table.qzv \
--m-sample-metadata-file sample-metadata.tsv
qiime feature-table tabulate-seqs \
--i-data rep-seqs.qza \
--o-visualization rep-seqs.qzv
qiime tools view table.qzv
qiime tools view rep-seqs.qzvtable

ref seq

5. 建树:用于多样性分析
# 多序列比对
qiime alignment mafft \
--i-sequences rep-seqs.qza \ --o-alignment aligned-rep-seqs.qza # 移除高变区
qiime alignment mask \
--i-alignment aligned-rep-seqs.qza \ --o-masked-alignment masked-aligned-rep-seqs.qza # 建树
qiime phylogeny fasttree \
--i-alignment masked-aligned-rep-seqs.qza \ --o-tree unrooted-tree.qza # 无根树转换为有根树
qiime phylogeny midpoint-root \
--i-tree unrooted-tree.qza \ --o-rooted-tree rooted-tree.qza6. Aplha多样性
# 指定重抽样的条数,使数据统一标准化,去除过低或过高的样品。标准化采用重抽样至序列一致。 --p-sampling-depth
qiime diversity core-metrics-phylogenetic \
--i-phylogeny rooted-tree.qza \
--i-table table.qza \
--p-sampling-depth 1109 \
--m-metadata-file sample-metadata.tsv \
--output-dir core-metrics-results
# 输出结果包括多种多样性结果,文件列表和解释如下:
# beta多样性bray_curtis距离矩阵 bray_curtis_distance_matrix.qza
# alpha多样性evenness(均匀度,考虑物种和丰度)指数 evenness_vector.qza
# alpha多样性faith_pd(考虑物种间进化关系)指数 faith_pd_vector.qza
# beta多样性jaccard距离矩阵 jaccard_distance_matrix.qza
# alpha多样性observed_otus(OTU数量)指数 observed_otus_vector.qza
# alpha多样性香农熵(考虑物种和丰度)指数 shannon_vector.qza
# beta多样性unweighted_unifrac距离矩阵,不考虑丰度 unweighted_unifrac_distance_matrix.qza
# beta多样性unweighted_unifrac距离矩阵,考虑丰度 weighted_unifrac_distance_matrix.qza
# 统计faith_pd算法Alpha多样性组间差异是否显著,输入多样性值、实验设计,输出统计结果
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/faith_pd_vector.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization core-metrics-results/faith-pd-group-significance.qzv
# 统计evenness组间差异是否显著
qiime diversity alpha-group-significance \
--i-alpha-diversity core-metrics-results/evenness_vector.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization core-metrics-results/evenness-group-significance.qzv
# 网页展示结果,只要是qzv的文件,均可用qiime tools view查看或在线https://view.qiime2.org/查看,以后不再赘述
qiime tools view core-metrics-results/faith-pd-group-significance.qzv
qiime tools view core-metrics-results/evenness-group-significance.qzvfaith-pd-group-significance.qzv结果


evenness-group-significance.qzv结果
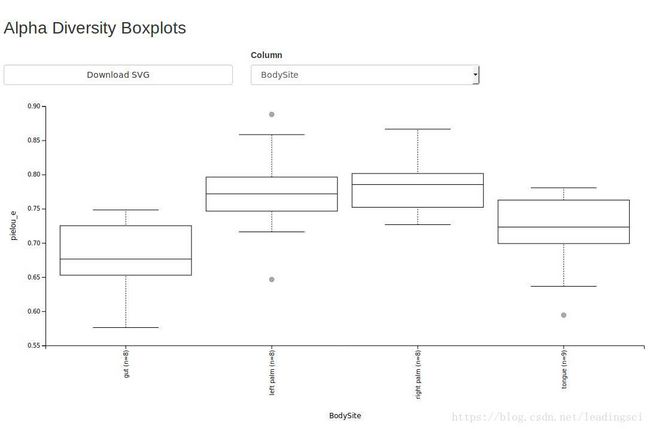

7.Beta多样性
# 按BodySite分组,统计unweighted_unifrace距离的组间是否有显著差异
qiime diversity beta-group-significance \
--i-distance-matrix core-metrics-results/unweighted_unifrac_distance_matrix.qza \ --m-metadata-file sample-metadata.tsv \ --m-metadata-column BodySite \ --o-visualization core-metrics-results/unweighted-unifrac-body-site-significance.qzv \ --p-pairwise
# 按Subject分组,统计unweighted_unifrace距离的组间是否有显著差异
qiime diversity beta-group-significance \
--i-distance-matrix core-metrics-results/unweighted_unifrac_distance_matrix.qza \ --m-metadata-file sample-metadata.tsv \ --m-metadata-column Subject \ --o-visualization core-metrics-results/unweighted-unifrac-subject-group-significance.qzv \ --p-pairwise
# 可视化三维展示unweighted-unifrac的主坐标轴分析
qiime emperor plot \
--i-pcoa core-metrics-results/unweighted_unifrac_pcoa_results.qza \ --m-metadata-file sample-metadata.tsv \ --p-custom-axis DaysSinceExperimentStart \ --o-visualization core-metrics-results/unweighted-unifrac-emperor.qzv
# 可视化三维展示unweighted_unifrac的主坐标轴分析
qiime emperor plot \
--i-pcoa core-metrics-results/unweighted_unifrac_pcoa_results.qza \ --m-metadata-file sample-metadata.tsv \ --p-custom-axes DaysSinceExperimentStart \ --o-visualization core-metrics-results/unweighted-unifrac-emperor-DaysSinceExperimentStart.qzv
# 可视化三维展示bray-curtis的主坐标轴分析
qiime emperor plot \
--i-pcoa core-metrics-results/bray_curtis_pcoa_results.qza \ --m-metadata-file sample-metadata.tsv \ --p-custom-axes DaysSinceExperimentStart \ --o-visualization core-metrics-results/bray-curtis-emperor-DaysSinceExperimentStart.qzv
# 网页展示结果,或下载在线查看
qiime tools view core-metrics-results/unweighted-unifrac-emperor-DaysSinceExperimentStart.qzv
qiime tools view core-metrics-results/bray-curtis-emperor-DaysSinceExperimentStart.qzvunweighted-unifrac

bray-curtis

8. Alpha rarefaction plotting
qiime diversity alpha-rarefaction \
--i-table table.qza \
--i-phylogeny rooted-tree.qza \
--p-max-depth 4000 \
--m-metadata-file sample-metadata.tsv \
--o-visualization alpha-rarefaction.qzv
qiime tools view alpha-rarefaction.qzv
9. 物种分类
# 下载物种注释
wget -O "gg-13-8-99-515-806-nb-classifier.qza" "https://data.qiime2.org/2018.4/common/gg-13-8-99-515-806-nb-classifier.qza"
# 物种分类
qiime feature-classifier classify-sklearn \
--i-classifier gg-13-8-99-515-806-nb-classifier.qza \
--i-reads rep-seqs.qza \
--o-classification taxonomy.qza
# 物种结果转换表格,可用于查看
qiime metadata tabulate \
--m-input-file taxonomy.qza \
--o-visualization taxonomy.qzv
# 展示taxonomy.qzv结果如下:
qiime tools view taxonomy.qzv
#Feature ID Taxonomy
#d12759fe8dda1d65fe9077cc1ca9cf28 k__Bacteria; p__Bacteroidetes; c__Flavobacteriia; o__Flavobacteriales; f__[Weeksellaceae]; g__Chryseobacterium; s__
#5ada68b9a081358e1a7d5f1d351e656a k__Bacteria; p__Fusobacteria; c__Fusobacteriia; o__Fusobacteriales; f__Leptotrichiaceae; g__Leptotrichia; s__
#d9095748835ade1b8914c5f57b6acbcf k__Bacteria; p__Proteobacteria; c__Gammaproteobacteria; o__Aeromonadales; f__Aeromonadaceae; g__Oceanisphaera; s__
# 物种分类柱状图
qiime taxa barplot \
--i-table table.qza \
--i-taxonomy taxonomy.qza \
--m-metadata-file sample-metadata.tsv \
--o-visualization taxa-bar-plots.qzv
qiime tools view taxa-bar-plots.qzv

10. Differential abundance testing with ANCOM——差异分析
# 只保留肠道样本
qiime feature-table filter-samples \
--i-table table.qza \ --m-metadata-file sample-metadata.tsv \ --p-where "BodySite='gut'" \ --o-filtered-table gut-table.qza
# # OTU表添加假count,因为ANCOM不允许有零
qiime composition add-pseudocount \
--i-table gut-table.qza \ --o-composition-table comp-gut-table.qza
# 采用ancon,按Subject分组进行差异统计
qiime composition ancom \
--i-table comp-gut-table.qza \ --m-metadata-file sample-metadata.tsv \ --m-metadata-column Subject \ --o-visualization ancom-Subject.qzv
# 查看结果
qiime tools view ancom-Subject.qzv
差异分类学级别分析:以按门水平合并再统计差异
# 按属水平进行合并,统计各门的总reads
qiime taxa collapse \
--i-table gut-table.qza \ --i-taxonomy taxonomy.qza \ --p-level 6 \ --o-collapsed-table gut-table-l6.qza
# 去除0
qiime composition add-pseudocount \
--i-table gut-table-l6.qza \ --o-composition-table comp-gut-table-l6.qza
# # 在属水平按取项目分类部分分析
qiime composition ancom \
--i-table comp-gut-table-l6.qza \ --m-metadata-file sample-metadata.tsv \ --m-metadata-column Subject \ --o-visualization l6-ancom-Subject.qzv
qiime tools view l6-ancom-Subject.qzv
图片解析有待补充
参考文章:
https://forum.qiime2.org/t/qiime2-chinese-manual/838