这是我听孟浩巍知乎LiveB站鲮鱼不会飞视频(转录组分析进阶课程)里的笔记哦~
本次知乎Live解决的问题:
转录组的建库与质量控制
- 人类基因组与转录组组成
- 转录组的2种建库策略
- 转录组的分链建库策略
- 转录组测序应用 single end 还是 pair end?
- 建库结果与测序结果的质控策略
转录组的比对与定量策略
- 常用比对策略介绍:bowtie, Tophat, STAR, HISAT
- 选择合适的参考基因组(hg19 / hg38)
- 确定基因表达量的若干策略:Raw Count, RSEM, FPKM, RPKM, TPM
- 基于FPKM的差异表达分析流程
- 基于 Raw Count 的差异表达分析流程
上机操作部分
使用 Python 与 R 搭建基于 Raw Count 的分析流程
导言
转录组分析的核心是找差异
找不同实验条件下、不同处理下、不同情况下基因表达量的差异、可变剪切的差异、基因融合的差异......
- 基因表达定量 ---> 差异表达基因(想要找差异表达基因那首先要确定基因的表达量)
- 可变剪切定量 ---> 可变剪切差异
所以说高通量分析转录组的核心就是选择一种更准确更快的方法对基因表达量进行定量从而找出差异基因。
测序之前要先建库,所以后续我们会讨论建库的方案。
拿到测序数据后,第一步先做质控;第二步把质控后的序列回帖到参考基因组,这步就需要考虑回帖的时候用什么软件什么策略更准确;第三步,回帖到参考基因组后怎样衡量基因表达量是高还是低;第四步,找可变剪切找差异表达基因。

在细胞运行过程当中RNA起到非常重要的作用,当然最主要的研究最多的是RNA的信息传递的作用。
我们的转录组分析其实就是分析这些RNA,其中最主要的是分析mRNA,但是除了mRNA之外的少量的其他种类RNA还有很多未知,更加值得进行进一步的挖掘。当然在众多种类的RNA中我们的mRNA是一个典型的代表,所以在进行转录组分析进阶的时候我们以mRNA为例。
人类基因组与转录组组成
在认识转录组之前首先要认识一下基因组,因为中心法则告诉我们RNA是来自于DNA的:
- 人类有46条染色体:22 * 2 + X + Y
- 人类基因组有两部分组成:细胞核基因组 和 线粒体基因组
- 我们常说的基因组是细胞核基因组大概有3.1Gb,线粒体基因组有16.6Kb
Human genome :3.1 Gb
chrM : 16.6Kb- 常用的基因有27000左右个(两万七千个常用的基因)
其中编码蛋白质的有两万个左右;能转录成有功能的RNA的基因有七千个左右
20000个左右蛋白基因
7000个左右RNA gene- 人类线粒体基因组中总共有37个基因,其中 22个tRNA基因;2个rRNA基因;13个编码蛋白的基因
RNA的种类与目前注释情况
左边这个饼图基本上就覆盖了所有注释好的RNA,只要做小RNA的分析基本上是不可能逃过这些数量的,这要超过这些数量就有预测的成分了。
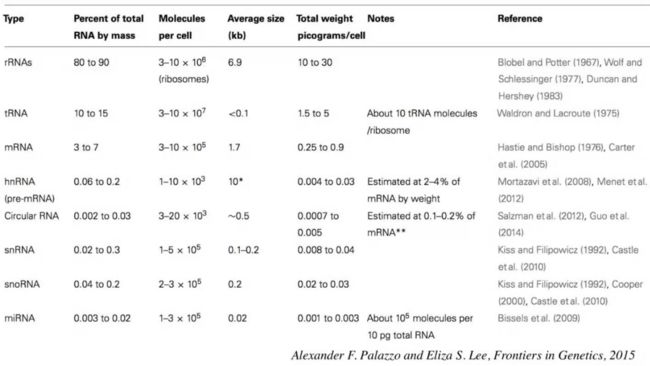
我们知道RNA有很多种类,每种RNA的含量大概是多少呢?下图的文献给我们做了非常好的总结。
rRNA也就是核糖体RNA占80%-90%,剩下的10%-20%绝大多数是tRNA,真正的我们所专注的 mRNA 只占5%左右。
我们提取出的RNA绝大部分是rRNA但我们真正关注的却是mRNA,此时我们应该富集,所以说转录组建库的核心就是将目标RNA比例提高。(常见的就是富集mRNA)
转录组建库的核心
- 转录组建库的核心是把目标RNA(最常见的是mRNA)的比例提高,然后进行扩增,使其浓度达到可以建库测序的要求。
- 单细胞转录组测序的核心,就是通过特殊的扩增方法,只扩增mRNA而不扩增其它种类的RNA,然后进行测序。
转录组的2种建库策略
- 第一种是正选择 —— polyA positive
- 第二种是负选择 —— rRNA minus
RNA-Seq的建库方法—怎样富集mRNA
polyA positive 建库方法
人的成熟的mRNA含有polyA尾巴,用Oligo dT(带有一段T序列的磁珠)标签去拉polyA尾巴,就会把mRNA从众多种类的RNA中拉出来,这种富集mRNA的方法就是polyA positive 建库方法。rRNA minus 建库方法
我们知道细胞中rRNA的含量最多(占到 80%-90%),那我们就想办法去除掉建库中最大的干扰因素rRNA,这种富集mRNA的方法就是 rRNA minus 建库方法。
接下来我们分别对这两种建库方法做详细介绍:
polyA positive 建库方法
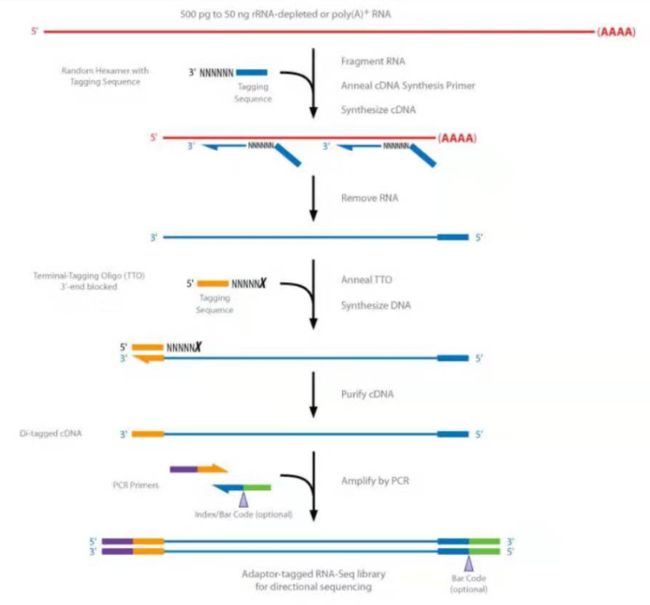
成熟的mRNA都有polyA尾巴,我们先用Oligo dT(带有一段T序列的磁珠)标签去识别polyA尾巴从而将mRNA富集出来,然后用 random 的 primer 对它进行扩增,此时我们就拿到了第一条链 是一条RNA、DNA杂合体,我们再用 random 的 primer 对第二条链进行扩增这时我们得到一个DNA和DNA的结构,这时我们再用末端修复酶把它补平为平末端然后在它 3' 末端加一个A,加完A后相当于把平末端变成一个黏性末端,之后加上adapter,就可以上机进行测序了。
rRNA minus 建库方法

第一步还是提出 total 的 RNA,我们有18S、28S、5.8S等这些不同种类的 rRNA 的标签,它特异性的结合 rRNA 的序列,这些标签上都有抗体方便我们特异性的识别并把它们取出,此时我们就去除了细胞中含量最多的RNA种类 — rRNA,一般为了保险起见,需要进行两次rRNA minus的去除,然后对剩下的RNA进行打断和建库。
两种建库方法的比较
第一种方法 — polyA positive 只拿成熟的mRNA进行建库,第二种方法 — rRNA minus 除了有不成熟的 mRNA 还有一些 tRNA、microRNA 等等
如果你只想研究成熟的mRNA那肯定选第一种建库策略,如果想研究一些其他种类的RNA或者RNA降解的一些问题,那必须选择第二种建库策略。
从两种建库策略我们可以看出,第二种建库策略涵盖的信息多,那为什么我们不都选择第二种呢?因为:第一,第二种策略需要更多的测序量;第二,第二种建库策略更贵,贵大概两倍左右。
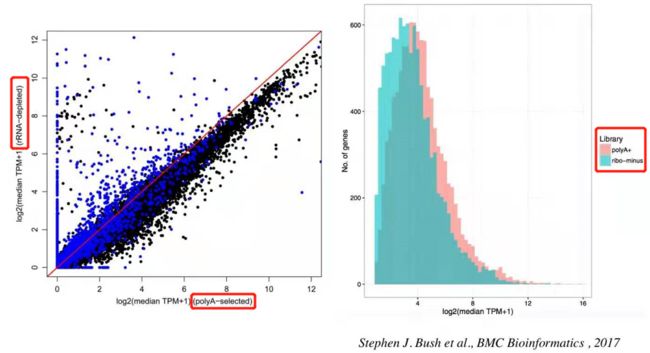
这篇paper对比了两种建库方法的基因表达量差异,发现了 rRNA minus 建库方法其原始数据更高一些,这也好理解,因为rRNA minus 建库时除了成熟的mRNA之外还有不成熟的mRNA(mRNA前体)和少量其他种类的RNA,所以最后测出的基因表达量整体比例会相对高一些。
两种建库策略比对到基因组后的差异
比对到参考基因组上,这两种比对策略会有什么不同呢?看这篇文章里面的图:
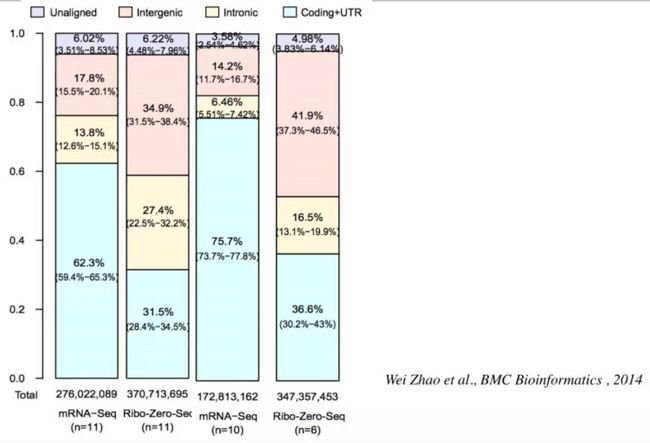
第一列和第三列是来自不同数据集,但用同一种建库策略(polyA positive)的结果,差别不大;第二列和第四列使用的是 rRNA minus 建库策略。
浅蓝色:Coding+UTR 表示能翻译成蛋白的 region
浅黄色:Intronic 内含子区
浅粉色:Intergenic 基因间 region
浅紫色:Unaligned 不确定的 region
第一种建库策略 (polyA positive) 即第一列和第三列中大部分的reads( 62.3%/75.7%)落在了Coding region,即能翻译成蛋白的区域;
第二种建库策略 (rRNA minus)即第二列和第四列中占比较多的是 Intergenic region以及 Intronic region,而在第一种建库策略中落在这两个区域的reads占比都很少,这是为什么呢?因为rRNA minus建库方法可以获得比较多的没有被剪切掉的Introne(内含子)的信息,用它来非常好的衡量 alternative splicing (可变剪切)。
整个基因组中能翻译成蛋白或有特定功能的序列我们叫做基因,基因在整个基因组中占比不超过5%,但是有一点我们必须清楚的知道,基因组绝大部分是可以被转录的只是除了那5%以外大部分序列并不行使功能,这就是为什么在使用rRNA minus建库的时候会有大部分的reads回帖到了 Intergenic region 即基因间区。
链特异性的转录组分析
在讲链特异性转录组分析之前回忆一下,之前是怎么建的库,以 polyA positive 为例。首先把带有polyA尾巴的mRNA富集出来,然后直接打断,再用random primer将mRNA进行反转录得到RNA、DNA杂合体,我们再用 random 的 primer 对第二条链进行扩增这时我们得到DNA和DNA的结构,当形成双链DNA的时候就丢失要传递的信息是来自于正链还是来自于反链?也就是在进行反转录的时候没有方向性使用的都是random primer,这就是我们平时所使用的链非特异性的测序,我们平时所测序的99%都是链非特异性的。
链非特异性测序会存在哪些问题呢?
我们在基因组分析的时候,特别是人类基因组,有一些基因是有overlap的,而且绝大多数的基因在转录的时候会出现偏差:比如有一段基因在正链在转录的时候不一定就老老实实的按照正链去转录,也会往反链转录一定的距离,这就会形成一些干扰。
解决这些干扰我们要使用链特异性的建库方法,链特异性建库方法最常用的是如图这三种,这三种中最常用的是中间那一种:
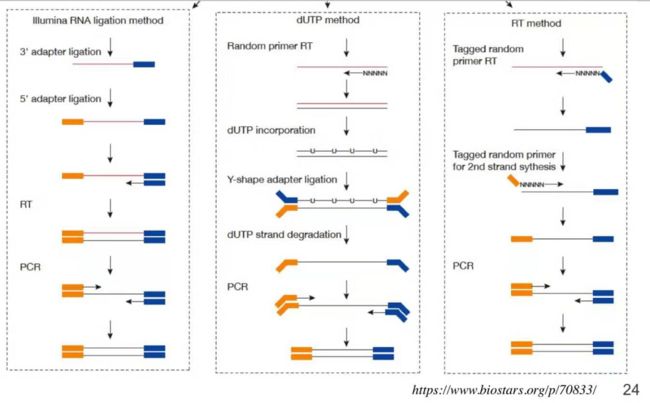
中间这种建库方法是基于dUTP的方法,首先不管用哪种方法进行富集,我们先得到了目标RNA(通常是mRNA),拿到RNA之后我们先用 random primer 进行反转,我们在合成第二条链的时候插入了U,我们知道DNA上是没有U的,这里我们人为的给它插入U,所以说我们合成的第二条链肯定带有U。它肯定来自于random primer 的转录而不是来自于最原始的那一条链,然后用专门能去掉U的酶,这种酶可以把含U的那条链全部切碎,这样的话就只剩下第一条链了,也就是反转录时的第一条最原始的链,我们用这些链进行建库。这时候左边的 adapter 和右边的 adapter 是有方向性的了,我们把这个信息保留下来,这样的话在进行建库测序的时候我们就可以知道它到底是来自于基因组的正链还是来自于基因组的负链。我们就通过这种方法解决了链的特异性的问题。当一个基因组正链也有基因反链也有基因的时候,某条序列只帖到反链没帖到正链,我们就可以说正链基因没有转录或者是转录水平非常低以至于检测不到。
我们举个例子:

ICAM4基因有001/002/003这三种transcript(转录本),它们是分布在正链的,它的反链还有一个基因叫做CTD,它在负链,如果不分链的情况下比对到正链的reads我们用蓝色表示,比对到负链的reads我们用绿色表示。不分链时(Nonstranded)就会发现结果既有蓝色又有绿色,这时我们不能说CTD基因不表达。
同样的样本我们用分链建库方法(Stranded)进行测序时就会发现,所有的序列全部回帖到了负链上,证明ICAM基因不表达。
单端测序(SE) VS 双端测序(PE)
测序的时候是选择单端测序还是双端测序?
答:能选择双端测序的时候一定选双端测序。
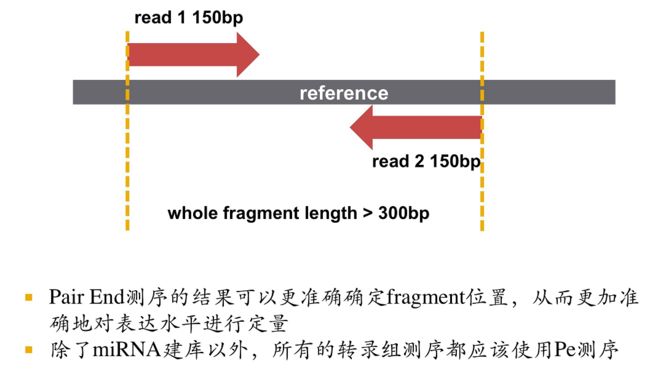
思考这样一个问题:人的基因组大概有3G,如果用
4^n 来覆盖大概有多长的序列能够unique map到基因组上呢?
大概是28-29bp,所有说那些短于28/29bp的序列,比对到参考基因组时会有好几个结果,也就是说这些短序列在基因组上找位置的时候能找到好多个。mapping到这么多位置上以至于我们不知道到底应该是哪个,这时候就应该延长测序长度。
illumina二代测序已经可以测到150bp,但是150bp map到一端还是不够的,所以我们在建库的时候打断成的片段尽量长一些,打断到500bp进行双端测序,上游也就是5' 端可以测到150bp,下游也就是reads2也可以测150bp,这样的话中间还空了200bp(500-150*2=200),在回帖到参考基因组上时,我们的reads1回帖到一个位置,reads2也回帖到一个位置,当这两个位置离的特别近大概在100/200bp的时候,我们就可以确定这是一个合理的回帖。这样我们就用了500bp的信息确定了这对reads 回帖到参考基因组的具体位置,也就是说用500bp的信息基本上实现了unique map。
所以说建议用双端测序(PE),这样可以使序列能够更好更准确的回帖到参考基因组上,基本上能够保证80%都是unique map。
但在测小RNA时是例外,比如microRNA,成熟的microRNA只有21-22bp,双端测150bp纯属浪费,单端测50bp就可以实现unique map。再比如最长的snoRNA也就300-400bp左右;成熟的tRNA 只有70bp; 成熟的snRNA也就70-80bp,所以说测这些小RNA(miRNA)的时候都不用双端150bp。
总结一下,针对mRNA建库无论是 polyA positive 还是 rRNA minus 也不管分不分链,一定要使用双端测序,小RNA建库的话要根据分析的内容确定是双端测序还是单端测序。
衡量提取RNA和建库的质量
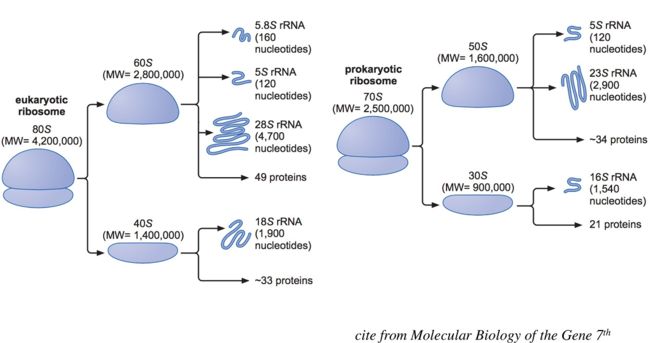
人体中绝大部分的RNA都是rRNA(核糖体RNA)占90%左右,人体的核糖体RNA主要分成两类,第一类,主要来源于细胞核,是根据细胞核里面染色体的序列转录并加工的;第二类,来自于线粒体中的两个rRNA基因转录而来的。针对第一类的rRNA主要分成:5.8S、5S、28S和18S。5.8S、28S和18S它们的量基本上是一致的,5S的量跟它们不太一样,因为5S会在整个基因组上散落分布。另外一种来自于线粒体上的有5S、23S和16S,整个rRNA最主要的、占比最多的是来源于细胞核的那四种(5.8S、5S、28S和18S)。
提取完 total RNA 要做一个电泳图,来判断提取的是否成功。
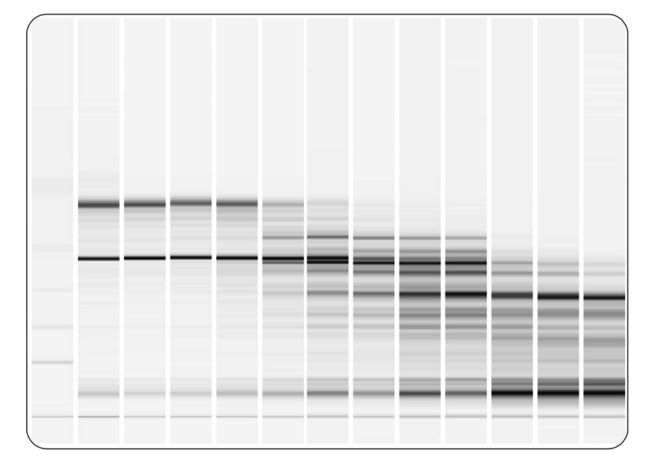
这个仪器在做出total RNA电泳图的同时还会计算出一个值来,通过这个值也能确定RNA提取的好不好。空气当中到处都是RNA酶,所以在提取RNA的时候,RNA特别容易降解,如果RNA讲解特别严重的话后面的流程就不用走了,测序结果也不准确。
跑胶的仪器可以测出 total RNA提取的质控标准 RIN(RNA Integrity Number)
RIN这个值就用来衡量total RNA提取的质量。RIN 10就代表完全没有降级,0就代表全部降解了。
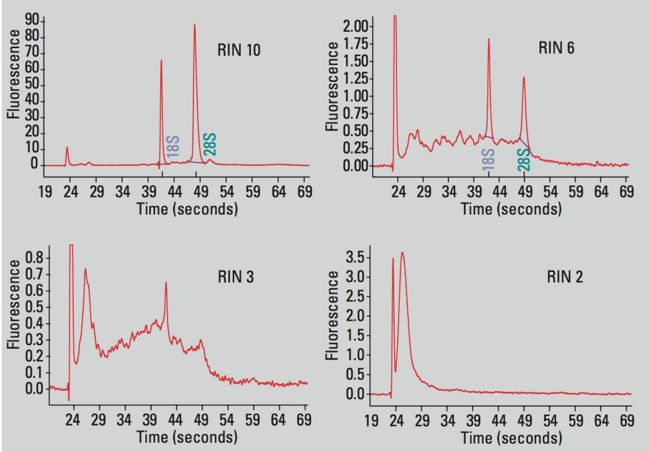
如上图所示,我们可以看到RIN 分别为 10、6、3、2 的图经验值告诉我们RIN 在7.5以上时,说明RNA提取的质量已经很好了。如果使用 rRNA minus这种建库策略RIN 值会稍微低一些,但是不能低于6.5;如果想做基因表达的分析的话RIN 值最低不能低于6,因为有一些病人的样本很珍贵,只能提一次,无法做平行那没办法只能硬着头皮往下做;如果是细胞类的实验,比如做植物类的这种可以多次重复的实验,尽量把RIN 值提高到7.5以上。 RIN 值在7.5及以上是可以做可变剪切分析的,如果达不到7.5是没办法做可变剪切的。
下面我们学习一下如何看这张电泳仪输出的total RNA提取的质量图。
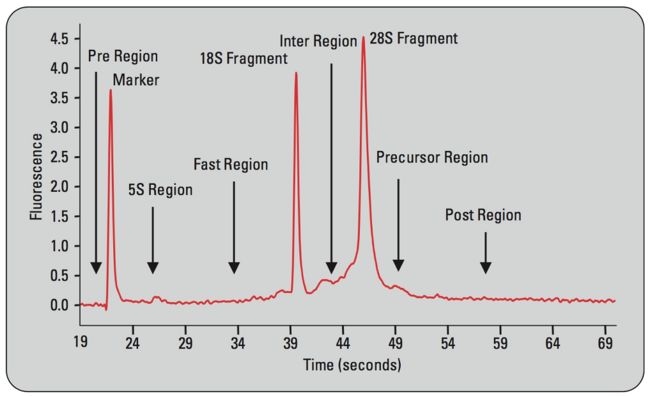
我们可以从上图中看到5S region 、18S region 、28S region 的peak,这些峰如果像marker的峰一样shark的话就比较好。
转录组的比对与定量策略
主要讲解拿到序列后怎样比对、怎样选择合适的参考基因组、用哪种参数定表达量?
怎样将提取来的RNA序列回帖到参考基因组,也就是怎样确定该RNA来源于哪个基因,也就是怎样进行转录组的mapping。
只讨论mRNA的情况下,一般来说有两种思路:
- 把测序的结果mapping到成熟的mRNA参考序列上;
优点:不用处理可变剪切的问题;
缺点:不能发现新的转录本 - 把测序结果mapping到参考基因组序列上;
优点:可以发现新的转录本,可以进行isoform层次的定量;
缺点:不能使用之前DNA mapping软件
一个基因有不同的isoform,不同的isoform的量是可以定下来的。
DNA mapping软件在mapping的时候没有 junction 的问题,只有indel、insertion、deletion,所以选择第二种比对策略需要新的mapping软件。
二代测序最实用的mapping算法是BWT算法,基于BWT算法的mapping软件:BWA、BWA-MEM、Bowtie、Bowtie2 ... ...
但是上述软件最大的问题是:针对DNA比对设计;完全没有考虑可变剪切的问题。
不能直接用于RNA的mapping
什么是可变剪切?

中心法则是从DNA到RNA,RNA到蛋白质,真实情况是在真核细胞里面,DNA转录成RNA时并不能全部转录成有用的mRNA,就会分成Exon(外显子)和 intron(内含子),成熟的mRNA都是由Exon(外显子)组成的,如图Exon1/2/3/4/5可以拼出一个成熟的mRNA;Exon1/2/4/5也可以拼出一个成熟的mRNA;Exon1/2/3/5也可以拼出一个成熟的mRNA,这三个成熟的mRNA分别可以翻译成对应的proteinA、proteinB、proteinC,但它们都是来自于同一个基因,对于这种来自同一个区域经过不同的剪切形成不同的mRNA最后翻译成不同的蛋白质这种就叫做不同的isoform,这种来自于同一个基因形成的三种不同的mRNA,我们叫做三种不同的转录本但是它们都是来自同一种基因。
在RNA比对的时候如果需要考虑可变剪切的问题,也就是说一条reads有可能来自于紧挨着的 Exon1、Exon2, 但是在回帖到基因组的时候 Exon1 和 Exon2 可能离的特别远中间会跨一个很大的junction,这种情况下怎么办呢?这个时候就需要这3种比对策略:
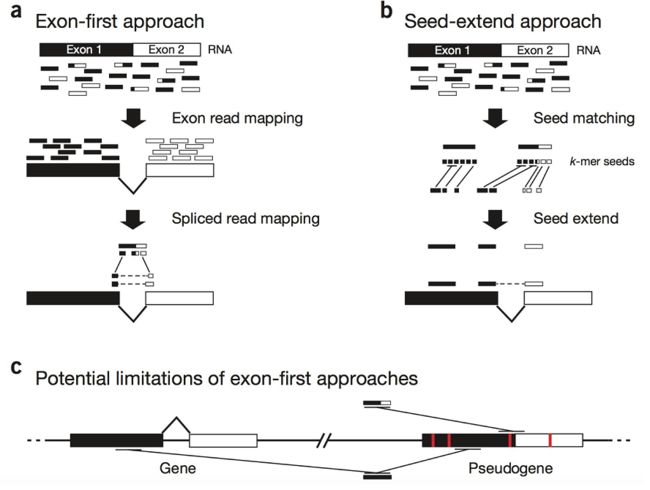
第一种比对策略是:Exon-first approach
基因组上哪些地方是Exon(外显子)哪些地方是 intron(内含子)我们是知道的。
把RNA序列完整的回帖到参考基因组上,能回帖上的就先回帖上,回帖不上的就按照一定的规则拆开,拆开后再分别去回帖,比如我们拆成两个部分一个回帖到了A位置,一个回帖到了B位置,A、B只要离的不太远且中间正好是一个intron(内含子)我们就认为这是一个合理的mapping。这种RNA比对方法就叫做Exon-first approach。
第二种比对策略是:Seed-extend approach
先把序列切的特别碎,这样每一段序列都相当于一个小种子(Seed),这种小种子在回帖在基因组时会map到非常非常多的位置,把小种子能map到的所有位置都找出来,对比Exon的样子把这些小序列能连在一起的连在一起,连不到一起的就认为是intron(内含子),这种RNA比对方法就叫做Seed-extend approach。
第三种比对策略是:Potential Limitation of exon-first approach
有点像第一种比对策略的改进版,在比对的时候一条序列如果它既能直接比对到基因组上,又能拆开之后比对到基因组上,比如,有一条reads正好能完全匹配到基因组的A位置,但是把如果把这条序列中间拆开一下,在中间有一个intron(内含子)的情况下却比对到了B位置,这种比对方法可以根据具体的需求选择是同时保留这两种情况,还是保留第一种或者保留第二种。
目前RNA-Seq的比对软件都是这三种比对策略当中的一种。
Tophat使用了第一种比对策略:
- 比对过程调用了Bowtie;
- 先比对能比对上的reads;
- 比对不上的reads根据可变剪切的方式拆开再比对;
-
最大的问题是,处理不好假基因(pseudogene)的问题
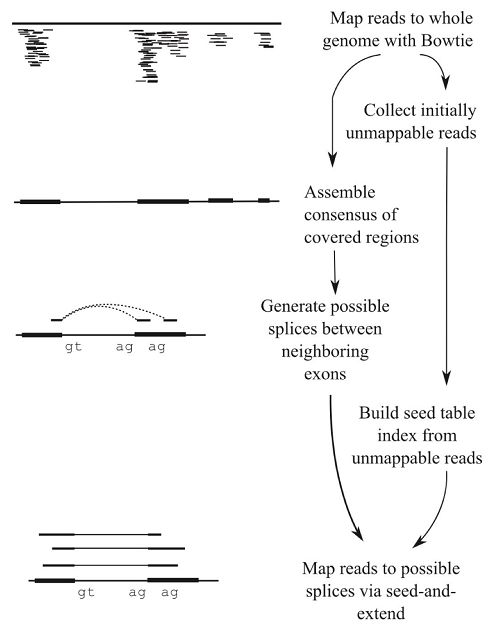 Tophat使用了第一种比对策略
Tophat使用了第一种比对策略
假基因(pseudogene)怎么来的呢?
正常情况下是DNA转录成RNA,RNA的Exon(外显子)再连到一起,intron(内含子)去掉,但是细胞里存在这样一个通路,可以把mRNA再插入到基因组上的一个位置,形成一个假基因(pseudogene),假基因(pseudogene)一般来说是不表达的,当A基因有一个假基因的时候我们叫 A' ,它的序列相当于A基因把所有Exon(外显子)都拼到一起了,这时候我有一条序列正好跨Exon(外显子)的junction,那么比对软件就会选在 A' 位置可以不用打开一个junction,直接就能比对到这儿,在基因A这个位置必须开一个intron(内含子)的junction才能比对到这儿,这个时候比对软件就可以很自然的把这条序列比对到A' 这个位置而不是A这个位置,这就是假基因所带来的问题。
Tophat2 主要改进的2个问题:
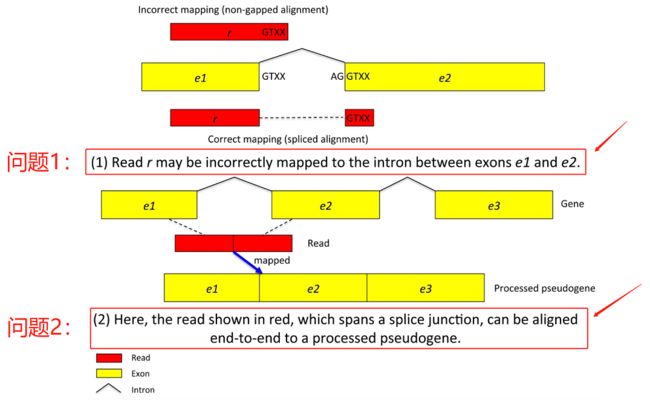

(1)是比对到转录组,这不重点介绍,因为转录组不是一个很复杂的问题,直接用bowtie比对也可以。
假如说有一条序列应该map到基因组,这该怎么实现的呢?
我们先看
spliced alignment(拼接比对),首先根据Alternative splicing(可变剪切)的 site 进行切分,切分完后再去比对这个过程分成了3-1、3-2、3-3、3-4、3-5这几种情况;
当遇到假基因(pseudogene)时会自动判断,然后根据权重分析是开一个junction,还是直接比对到参基因组,这样就通过权重考虑解决了假基因的问题;
另一个问题是,当
intron(内含子)有一小段和序列延伸出来那部分完全匹配上的话怎么办?
这种时候能开junction就尽量开junction,这样比对的整体效果会好很多。
整体来说,Tophat2比对的效果还是不错的,但是有一个最大得到问题就是占用资源太多,而且比对的速度太慢,因为它在比对的时候存在一个调用的过程 —— Tophat调用的是bowtie;Tophat2调用的是bowtie2
所以说,同一伙儿人又搞了一个HISAT软件,它主要是一个Tophat2升级的版本提高了index的构建方式,从而达到占用资源小,比对速度快的目的。在进行转录组分析时推荐使用HISAT软件进行比对
还有一个常用的比对软件是STAR,它的优势是快,缺点是占用内存比较大,我们知道人类基因组一共才有3G,但STAR在mapping的时候需要28-32G的内存。
STAR软件的比对策略
使用的是上面介绍的第二种比对策略
- STAR的优势在于快,能够快速mapping;
-
缺点在于占用内存巨大,human的mapping需要28-32G的内存
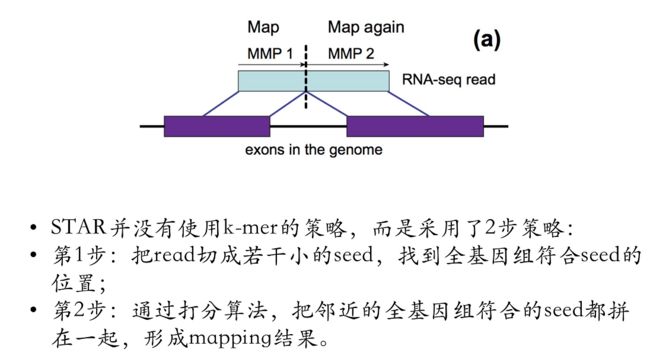 STAR软件的比对策略
STAR软件的比对策略
为了能够快速mapping,STAR软件把全部的seed提前做好了,index会提前读进去,所以说内存占用非常大。
许多比对软件,比如:Tophat、Tophat2、HISAT 它们的核心算法都是BWT算法
BWT的算法推导与核心原理:孟浩巍知乎文章链接
RPKM VS FPKM
先理解一下所谓的 raw count 是什么?就是样本中比对到某个基因的 reads count
比如某个测序的序列比对到基因的Exon(外显子)上,
样本一中 a基因比对上了1000条reads,b基因比对上了2000条reads
样本二中 a基因比对到了2000条reads,b基因比对到了4000条reads
这时,是否就可以认为:
1、样本内比较,b基因的表达量就比 a 基因高
2、样本间比较,a/b两个基因的表达量样本二比样本一高出两倍
答案肯定是不行的
我们看下面这张图,有四个基因分别是1/2/3/4:
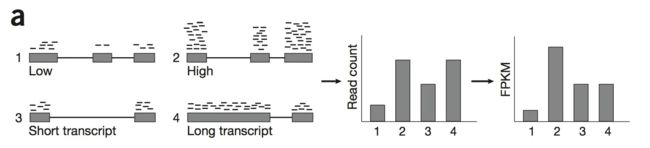
如果我们只单单看 reads count,那很明显,1/3基因表达量较低,2/4基因表达量较高。4号基因和2号基因有同样的基因map到,但是转录本的长度不一样,所以需要先矫正转录本长度;样本间比较的话也是需要矫正的,样本一 测了1兆reads,样本二 测了10兆reads ,那样本二的基因表达量就会比样本一的高,综上,我们需要对我们的
raw count 进行
normaliza 。
normaliza 最重要的两个因素,一个是转录本的长度(或者叫Exon(外显子)的长度);另一个就是测序量
RPKM / FPKM 是一对儿概念

RPKM翻译成人话就是说,每测1兆reads,在基因a上平均每1k的Exon(外显子)有几条reads map到这里。
FPKM里的F是指
Fragment ,只有双端测序才有
Fragment ,双端测序中一对叫一个
Fragment ,FPKM翻译成人话就是每测1兆reads pair 基因a的位置上,平均1k的Exon(外显子)上map回来多少个
Fragment。
所以说针对illumina二代测序 pair-end(双末端) 的数据,FPKM是该基因RPKM数值的二分之一,换句话说就是,RPKM = FPKM * 2
RPKM / FPKM就相当于对测序量和基因Exon(外显子)的长度进行了矫正。
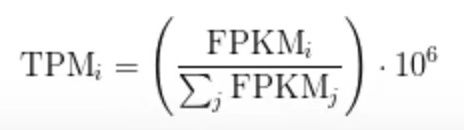
TPM中T指的是transcript(转录本),翻译成人话就是每测1兆reads,每一条转录本上有多上条reads map上,相当于FPKM的小修小改。
在计算FPKM的时,比如有两个样本测序量不一样那不同基因的表达量有可以一样也有可能不一样,但FPKM的总体加和往往是不一样的,这就会让我们觉着在比对的时候会出现问题,TPM就是为了应对这样的问题而出现的,比如说我们算基因 i 的TPM,根据公式我们首先必须算它的FPKM,它的FPKM占整个基因组的FPKM的百分之多少再乘以 10^6 就是TPM了,所以说TPM的本质是一个百分数,一个RNA-Seq的TPM一定是10^6 ,TPM的优势在于它可以很好的衡量样本之间表达量差异的情况,因为它把整个样本进行normaliza 了,保证了各样本TPM的加和是一致的是10^6,也就是这样的原因TPM得到了越来越多的推崇。

RSEM主要是用了机器学习的办法来估基因的 reads count 有多少,RSEM数值上相当于对测序量和基因Exon(外显子)的长度进行了矫正,与传统的FPKM不太一样的是,传统的FPKM也是进行了矫正,但FPKM是直接除以了Exon的长度,而RSEM认为这样并不能完全解决问题,应该把它看成一个概率模型估出来一个修正的值,这个处理流程是一个很复杂的流程:
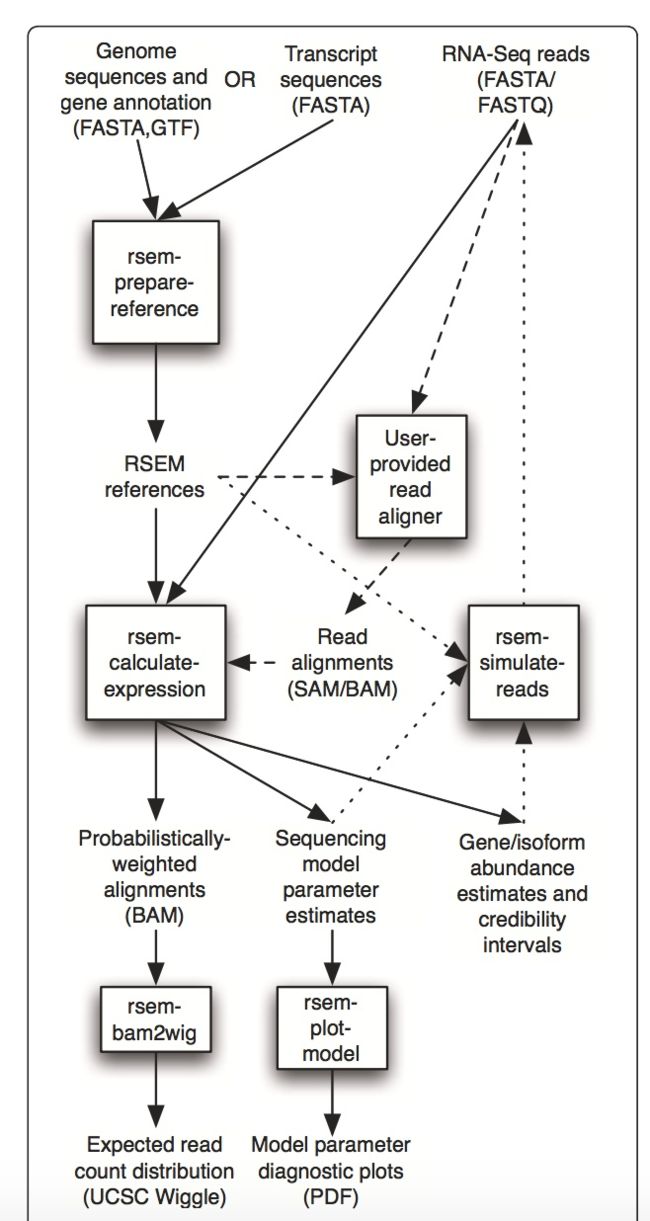
RSEM主要是用了机器学习的办法来估基因的 reads count 有多少,在估计的时候使用了EM算法,R是
RNA,S是
seek,E是
expectation,M是
maximization
EM的意思就是期望最大化,所以RSEM就是用了EM算法来估计基因真正的、合理的reads 数量。
总体来说,RPKM没什么人用了,因为现在基本上都是 pair-end 测序,FPKM现在是主流,但是FPKM不如TPM方便,TCGA数据库里面给了很多的表达量都是RSEM。
RSEM整个的评估号称要比FPKM要稳定和准确,但是RSEM在理解上不是很直观。
TPM是一个比较好的标准,因为它又矫正了长度又矫正了测序量而且还能保证每个sample总数是固定的。
上机操作部分
- RNA-Seq的本质是什么?
- RNA-Seq寻找差异基因是为了干什么?
- Raw Count 是什么东西?怎么算?
- FPKM/TPM 是什么东西?怎么算?
- Cuffdiff的原理
- DESeq2的原理
转录组在整个NGS 中是一个核心的位置,它启到承上启下的作用,承上它是对DNA进行了转录,启下就是必须有蛋白才能行使功能,没有RNA是不可能有蛋白质的
RNA-Seq的本质是对RNA进行定量!!!
思考:对RNA进行定量了怎么就有意义了?RNA定量定的是绝对量还是相对量呢?
答:RNA-Seq定量定的是相对的量
为什么要去找差异基因?
- DNA ---> RNA ---> 蛋白质 ---> 功能
- 要实现一个具体的功能,蛋白的含量大概率需要变化,而蛋白含量的变化要形成一个稳态,RNA水平大概率会变
- 综上,寻找差异基因的本质是通过RNA变化来推测蛋白的变化
Raw Count 是什么东西?
我们已经知道了RNA-Seq是为了找差异表达基因也就是 DEG differencial expression genes
找差异那一定得有至少两个来比较,也就是说在寻找差异表达基因的时候一定得有一个 input1 和 input2
GeneA:input1 - reads count --> 数一下在input1中有多少reads map到GeneA;
GeneA:input2 - reads count --> 数一下在input2中有多少reads map到GeneA
fold change = input2 - reads count / input1 - reads count
我们把得到的这两个数一除,就得到 fold change(倍数变化)
FC = 1.5 ? p-value ?
FC = 1.1 ? p-value ?
然后进行一个统计检验,告诉我们 fold change 等于1.5时,差异是否显著,p-value是多少;fold change 等于1.1时,差异是否显著,p-value是多少。
也就是说,对于GeneA我们想知道它的表达量是升高了还是降低了,我们只要数在对照组里面(input1)有多少条reads回帖到了GeneA,然后在实验组里面(input2)数有多少条reads回帖到了GeneA,然后算一个fold change ( input2 - reads count / input1 - reads count),等到的FC再进行统计检验。
在这个过程中,值得注意的两个问题是:
- 一、测序量是否一致
- 二、2000 --> 2500 VS 200 --> 250 这两个变化到底哪个更显著?
这两个问题在RNA-Seq中属于比较棘手的问题。
问题1,测序量的问题其实是很好解决的,直接矫正测序量就可以了,也就是我们常说的 normalization 常用的有这三种矫正方法:
- quantile
- genometry
- traditional
问题2,怎样去衡量,哪个才是真正显著的?
基本假设!!!!:
- 大部分的基因表达量不变;
- 高表达的基因表达量不变
Why?回答这个问题我们一定要明白 p-value 是怎么算出来的?
我们首先得有一个0假设-->两者没有差异,再来一个1假设-->两者有差异,然后算出一个统计量x ,给出一个置信区间阿尔法α,这种条件下肯定H0或者否定H1
H0:两者没有差异
H1:两者有差异
统计量x,α = 0.05,肯定H0或者否定H1
input1(对照组)和input2(实验组)在进行统计检验的时候,认为GeneA在两组条件下表达没有差异,两个分布应该是同一个整体分布,在同一个整体分布的时候观察到在input1总体中(对照组)reads count 为2000的情况下p-value为多少?同时观察在input2总体中(实验组)reads count 为2500的情况下p-value为多少?
假如 p-value = 0.001 即单次试验 p-value<0.05 ,说明这是一个小概率事件,小概率事件在单次实验中认为不发生,但它发生了,所以我们认为我们的0假设不对,所以两者有差异。
知道了这个,我们再回推回去,为什么大部分的基因表达量不变?就是因为我们的0假设,大部分的基因表达量不能变如果变的话,我们所有的统计没有办法操作。
在捋一下p-value的事儿:
H0是认为两者没有差异;H1是认为两者有差异,构造一个统计量在input1中观察到reads count是2000,在input2中观察到reads count 是2500,那么观测2000和2500的概率是p-value=0.001
由于p-value<0.05,那么这就是一个小概率事件,小概率事件单次实验不发生,但这里却发生了,因此否定H0,所以有差异。
解决第二个问题:为什么高表达的基因不能变?
假如说基因组就有 gene A ,B,C
总共测了200条reads,gene A有150条;geneB 和geneC 各25条 这是input1;在input2中同样只有gene A ,B,C,同样测了200条reads,当高表达量基因 geneA降低到100条,gene B C 没有变化时,
客观条件下应该是这种情况:
input1:200 = 150 + 25 + 25
input2:150 = 100 + 25 + 25
但,实际测的时候会出现这种情况:
input1:200 = 150 + 25 + 25
input2:200 = 130 + 35 + 35
实际上高表达基因 gene A 从150 变成了100 发生了显著变化,gene B C 没有发生显著变化;但是实际测的情况确实基因 A 从150 变成了 130 并没有发生显著变化,而gene B C 从25 变到 35 发生了显著变化
所以说,当一个高表达基因发生显著变化的时候,它会把其他基因拉跑偏,所以说我们在进行统计检验的时候一定要遵循基本假设:大部分的基因表达量不变;高表达的基因表达量不变。
那么,问题来了,如果高表达的基因表达量变了怎么办?如果大部分的基因表达量发生了变化怎么办?
解决方案:
- 第一,掺入
spike-in(适用于解决1/2假设不成立时)
人工掺入一个我们已知量的东西,变成绝对定量 - 第二,使用
house keeping gene进行校正 (适用于解决2假设不成立时)
house keeping gene是维持细胞基本代谢所需要的基因,而且细胞和细胞之间house keeping gene的表达量不太变。
FPKM的本质是什么?
FPKM本质是对测序深度,基因长度做了矫正
FPKM =(reads count / exon length / total reads)* 10^9
最后乘以10^9 是为了单位好看一些
TPM类似
TPM = (FPKM / total FPKM)* 10^6

所以说,
FPKM和
TPM都对测序深度和基因长度做了矫正,而 Raw count 不对基因长度做矫正只对测序量进行矫正。
那么,
FPKM和
Raw count到底哪个好哪个不好呢?这就公说公有理婆说婆有理了

cuffdiff pipeline:
FASTQ-reads --> mapping (HISAT2,tophat2)--> BAM --> cuffdiff --> DEGs(FPKM)

DESeq2 pipeline:
FASTQ-reads --> mapping (HISAT2,tophat2)--> BAM --> Gene Raw Count --> find DEGs in R (No FPKM)

泊松分布与负二项分布

泊松分布就一个参数叫 lambda,期望(E),和方差(Var)都是 lambda。用来衡量不太容易发生的事儿:
比如,小茗同学有一定概率会被雷劈,那么他这一生当中被雷劈的次数基本上是符合泊松分布的。到我们的基因组上来说就是,有多少条
reads 能够 map 到
Gene A上,在最早的时候我们认为它是符合泊松分布的,后来我们就认为它符合负二项分布。
什么是负二项分布?
有两盒火柴分别有A根火柴和B根火柴,把A盒火柴放到左口袋里,把B盒火柴放到右口袋里,每次用火柴的时候随机从左口袋或者右口袋取一根,直到有第一盒火柴用完的时候,那用火柴的次数 x,一定满足:sum(A, B) >= x >= min(A, B)
再次举例说明:
一袋小球里面有黑球和白球,黑球有a个,白球有b个,每次从袋子里随机抽一个小球,需要抽多少次才能把其中一种颜色的球抽完?这个问题就和我们基因组的问题很接近了,我们基因组可以分成两个部分:基因 和 非基因。对于任意一个基因 gene1,基因上就是:gene1,non-gene1,测序比对后就会发现有一些reads会map到gene1的位置,那么能map到gene1位置的事件就近似服从负二项分布。因为就相当于从整体里面进行抽样,我认为能map到gene1就是从整体里面去抽样正好把gene1抽出来了,抽完之后reads再回帖回来。
负二项分布和泊松分布其实差不太多,在次数比较大的时候这两个没啥区别,次数比较小的时候负二项分布的离散性更低一些。基本上我们现在做的基因组的统计检验都是用负二项分布。
学习DESeq2包进行差异表达分析
使用DESeq2包进行差异表达分析首先需要计算
Raw reads Count
计算gene的Raw reads Count的方法
方法1. 使用知乎Live主—孟浩巍的代码,或者根据代码修改自己的需求;
优点:在R中完成操作,支持多线程;
缺点:内存占用比较大;方法2. 使用htseq-count程序
优点:节省内存
缺点:不够优雅,需要对读取进来的内容再做处理
方法1,2本质没有差别,我们以方法1为例子进行演示
将孟浩巍写的计算 Raw reads Count 的R代码文件Count_RNASeq.R放到服务器上(R代码解释后面讲)
我们在服务器的命令行输入 Rscript Count_RNASeq.R 就会告诉我们命令格式:
$ Rscript Count_RNASeq.R
Loading required package: magrittr
Count_RNASeq.R Usage like:
----------------------------------------------------------------------------------------------------
Rscript Count_RNASeq.R --input XXX --label XXX --type XXX --threads XXX --output XXX
----------------------------------------------------------------------------------------------------
--input : input bam file list, format like bam1,bam2,bam3...
--label : the same length as --input, format likt label1,label2,label3...
--type : the sample type ,the same length as --input, format likt ctrl,ctrl,ko,ko...
--threads : the CPU limit
--output : name of RData that contain SummarizedExperiment.obj
Error: Input parameters error!
Execution halted
单看不太容易看明白其实举个例子就很简单,比如,我们有两个分组 WT、KO,那么--input --label --type --threads --output 这些命令后面的文件格式按照上面给的要求写好就是这个样子:
--input ./wt.rep1.bam,./wt.rep2.bam,./wt.rep3.bam,./ko.rep1.bam,./ko.rep2.bam,./ko.rep3.bam,
--label WT.rep1,WT.rep2,WT.rep3,KO.rep1,KO.rep2,KO.rep3
--type WT,WT,WT,KO,KO,KO
--threads 6
--output ./RNA_raw_count.RData
注意,--input 和--label后面的文件是一一对应的; --threads是指定用多少个核,我们这里有6个文件,所以指定的是6个核,多了也没用少了浪费时间; --output 最后会保存成R语言能够读成的格式,自己起个名字后缀加上.RData就行。
各个参数写好之后还要调整一下(中间可以有空格但不能换行):
Rscript Count_RNASeq.R --input ./wt.rep1.bam,./wt.rep2.bam,./wt.rep3.bam,./ko.rep1.bam,./ko.rep2.bam,./ko.rep3.bam, --label WT.rep1,WT.rep2,WT.rep3,KO.rep1,KO.rep2,KO.rep3 --type WT,WT,WT,KO,KO,KO --threads 6 --output ./RNA_raw_count.RData
这样我们就可以在服务器上运行计算gene的Raw reads Count的程序了,为什么不直接在RStudio上直接运行,还要这么费劲的将R代码整到服务器上,因为个人电脑带不起来呀!!!
我们把Count_RNASeq.R 文件(计算gene的Raw reads Count的代码文件)里的R代码理解一下
打印刚刚的说明文档代码:
# help function
my_help <- function(){
cat("Count_RNASeq.R Usage like:\n")
cat("----------------------------------------------------------------------------------------------------\n")
cat("Rscript Count_RNASeq.R --input XXX --label XXX --type XXX --threads XXX --output XXX\n")
cat("----------------------------------------------------------------------------------------------------\n")
cat("\t--input : input bam file list, format like bam1,bam2,bam3...\n")
cat("\t--label : the same length as --input, format likt label1,label2,label3...\n")
cat("\t--type : the sample type ,the same length as --input, format likt ctrl,ctrl,ko,ko...\n")
cat("\t--threads : the CPU limit \n")
cat("\t--output : name of RData that contain SummarizedExperiment.obj\n")
}
读参数,输入的参数是通过下面这样的方式读进来的:
# 1. check args length
args = commandArgs()
args.length = length(args)
if(args.length != (5 + 5 * 2)){
my_help()
stop("Input parameters error!")
}
检查是否有这些:--input --label --type --threads --output 指定的参数,如果没有就停止,然后打印说明文档(my_help())并报出错误提示:Input parameters error!:
# 3.check if all parameter in the list
parameter_list = c("--input","--label","--type","--threads","--output")
if(! all(parameter_list %in% names(args.list))){
my_help()
stop("Input parameters keys error!")
}
如果输入的都没有问题,就会把变量都定义好,告诉你INPUT_BAM INPUT_LABEL INPUT_TYPE CPU_THREADS OUTPUT_FILE 都是些啥:
# 4. reads parameters
INPUT_BAM = strsplit(args.list[["--input"]],split=",") %>% unlist()
INPUT_LABEL = strsplit(args.list[["--label"]],split=",") %>% unlist()
INPUT_TYPE = strsplit(args.list[["--type"]],split=",") %>% unlist()
CPU_THREADS = strsplit(args.list[["--threads"]],split=",") %>% unlist() %>% as.integer()
OUTPUT_FILE = strsplit(args.list[["--output"]],split=",") %>% unlist() %>% path.expand()
OUTPUT_DIR = dirname(OUTPUT_FILE)
OUTPUT_BASENAME = basename(OUTPUT_FILE)
然后检查BAM文件是否存在:
# 5. check if BAM file exist
if(all(file.exists(INPUT_BAM))){
print("All file checked exsit!")
}else{
stop(sprintf("File: '%s' Not Exist!",INPUT_BAM[!file.exists(INPUT_BAM)]))
}
BAM文件是比对的结果文件,里面存放了哪些reads比对到了基因组的哪些位置,我们就是要用BAM文件来计算Raw reads Count,所以必须检查是否存在BAMwe文件。
如果BAM文件不存在会报:.......Not Exist!,如果所有的文件都存在所有的设置没有问题,下面就开始正式的把这些包都载入:
######################################################################################
# required packages
######################################################################################
require(BiocParallel)
require(GenomicFeatures)
require(GenomicAlignments)
require(Rsamtools)
require(TxDb.Hsapiens.UCSC.hg19.knownGene)
这个包BiocParallel是R里面并行的一个包;这个包GenomicFeatures是做GTF分析必须得用的用来真正数 reads count 的;这个包Rsamtools是打开BAM文件用的;这个包TxDb.Hsapiens.UCSC.hg19.knownGene是人的参考基因组转录组信息。
把input_bam信息读进来:
input_bamfiles = Rsamtools::BamFileList(INPUT_BAM)
读进来之后把 bam 文件标上 label 就是告诉你每个 bam 是什么:
names(input_bamfiles) = INPUT_LABEL
这两部整合起来就是:
######################################################################################
# read BAM file
######################################################################################
input_bamfiles = Rsamtools::BamFileList(INPUT_BAM)
names(input_bamfiles) = INPUT_LABEL
读入人的GTF文件:
######################################################################################
# load GTF file
######################################################################################
print("Loading GTF file...")
gene_range = GenomicFeatures::exonsBy(TxDb.Hsapiens.UCSC.hg19.knownGene,by="gene")
print("Done!")
一切准备就绪我们就开始counting了:
######################################################################################
# counting gene reads
######################################################################################
print("counting...")
# BiocParallel::register(SerialParam())
BiocParallel::register(MulticoreParam(workers=CPU_THREADS))
SummarizedExperiment.obj <- GenomicAlignments::summarizeOverlaps(features=gene_range,
reads=input_bamfiles,
mode="Union",
singleEnd=FALSE,
ignore.strand=TRUE,
fragments=TRUE )
print("counting...Done!")
其中 mode 这个参数有三个选项,这里用的是mode="Union"默认情况下都是Union模式,另外两个分别是:IntersectionStrict 和 IntersectionNotEmpty
此时,count完成,保存有用的信息成为一个对象SummarizedExperiment.obj
并按照指定的输出位置输出文件:
######################################################################################
# save data
######################################################################################
# make sample table
sample_table = DataFrame(case_label = INPUT_LABEL,
case_type = INPUT_TYPE,
bam_path = INPUT_BAM)
colData(SummarizedExperiment.obj) = sample_table
# save SummarizedExperiment.obj
save(file=OUTPUT_FILE,list="SummarizedExperiment.obj")
print("Save R image... Done!")
rm(list=ls())
print("Clear R env... Done!")
最后清空所有变量,操作完成~
DESeq2包进行差异表达分析
计算完gene的raw reads count得到结果文件SummarizedExperiment.obj后我们就可以用DESeq2包进行差异表达分析了。
读入上一步计算raw reads count的结果文件 —SummarizedExperiment.obj:
#### 读取上一步的计算结果文件 — SummarizedExperiment.obj
load(file="./ReadsCount.RData")
SummarizedExperiment.obj
colData(SummarizedExperiment.obj)
我们看下上面代码在R里的运行结果:
> load(file="./ReadsCount.RData")
> SummarizedExperiment.obj
class: RangedSummarizedExperiment
dim: 23459 4
metadata(0):
assays(1): counts
rownames(23459): 1 10 ... 9994 9997
rowData names(0):
colnames: NULL
colData names(2): case_label case_type
> colData(SummarizedExperiment.obj)
DataFrame with 4 rows and 2 columns
case_label case_type
1 WT.rep1 WT
2 WT.rep2 WT
3 KO.rep1 KO
4 KO.rep2 KO
>
colData(SummarizedExperiment.obj)运行结果告诉了我们SummarizedExperiment.obj文件里的保存信息。我们可以看到case_type那一列是样本信息分别是 WT 和 KO。
结果读进来后第一步就应该构建 DESeq 的 DataSet,因为我们这里只需要区分WT 和 KO,所以我们这里面的参数design只需要按照case_type去区分就可以了,即:design = ~case_type
#### 构建DESeqDataSet
DES_set = DESeqDataSet(SummarizedExperiment.obj,design = ~case_type)
做差异表达分析就是将上一步构建好的DESeq的DataSet — DES_set,扔到函数DESeq中即可:
#### 进行差异表达分析
DEG_set.run = DESeq(DES_set)
#### 输出结果
DEG_set.result = results(DEG_set.run)
DEG_set.result
#### 对结果进行统计
summary(DEG_set.result)
#### 将结果保存成data.frame
DEG_set.result.df = as.data.frame(DEG_set.result)
DEG_set.result.df就是最终的结果文件了,我们打开看一下
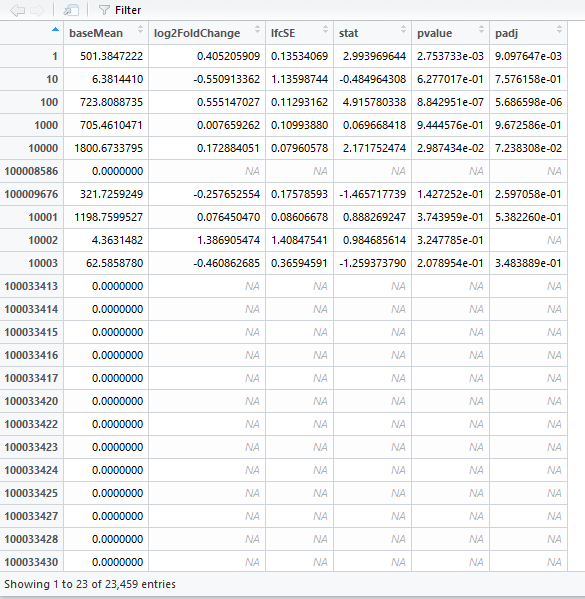
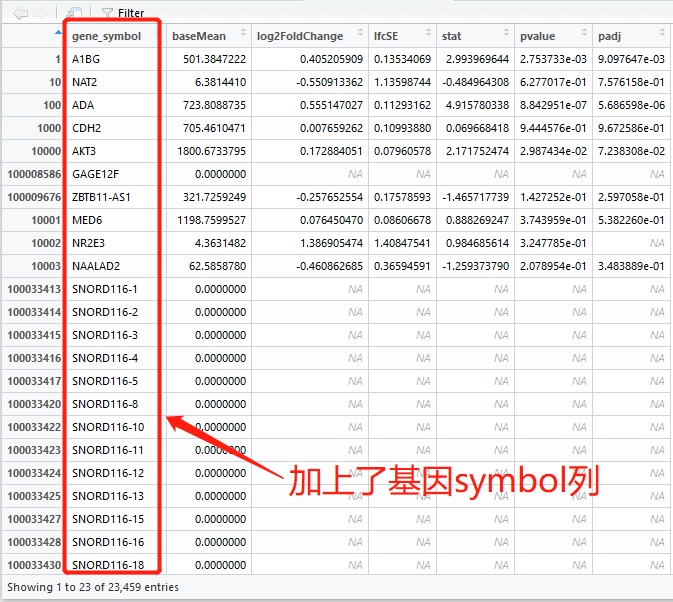
我们可以看到列有
log2FoldChange还有
pvalue,我们一般用
padj。
最左边那一列是基因ID编号,下面我们把它改成
gene_symbol(基因symbol):
library(org.Hs.eg.db)
columns(org.Hs.eg.db)
gene_symbol = mapIds(x=org.Hs.eg.db,
keys = rownames(DEG_set.result.df),
keytype = "ENTREZID",
column = "SYMBOL")
我们使用的是mapIds这个函数来准换的,其中各参数解释:
-
x是基因ID和基因symbol对应的数据库,人的就是org.Hs.eg.db; -
keys来指定想要转换的数据,这里就是基因ID号即结果文件DEG_set.result.df的行名(rownames); -
keytype是想要转换的数据类型,这里是基因ID号学名就是ENTREZID; -
column来指定我们想把它准换成的类型,这里是把基因ID准换成对应的基因名,学名的就是基因SYMBOL。
DEG_set.result.df.fix = data.frame(gene_symbol,DEG_set.result.df)
这行代码就是将转换好的gene_symbol加到原来的结果文件DEG_set.result.df中,形成新的带有基因symbol的结果文件 —— DEG_set.result.df.fix
加餐:
house keeping gene矫正
先搞到house keeping gene list

这篇文章把当时市面上所有的RNA-Seq数据全部都分析了一遍,找到了大概两千多个他们认为的
house keeping gene,还推荐了一些适合用做
qPCR 验证和当内参的
house keeping gene,这个基因 list 从网上直接搜就可以了:

list搞到之后,使用
RUVSeq包进行验证,这个包的说明书写的非常非常详细,看包操作就行。
如果有多个
input是不是两两比较?
如果input不是很多那我们就进行两两比较,如果比较多有十几个那就用cuffnorm,不做显著性差异了,用cuffnorm把大家都normalization一下 看一下某一类的基因在这十几个样本里面表达量的升高和降低,这是第一个分析,第二个分析是用加权共表达网络分析WGCNA,这也是比较常用的。
输入的时候输入 raw reads count 、FPKM、RSEM、TPM 都可以,但一定要保证输入是一致的,个人建议TPM好一些。