topGO手册中的实例实现
手册地址:http://bioconductor.uib.no/2.7/bioc/vignettes/topGO/inst/doc/topGO.pdf
快速入门部分可以参考:https://rpubs.com/aemoore62/TopGo_colMap_Func_Troubleshoot
1. 导入基因和注释数据
用library(topGO)导入topGO包后,会自动创建三个环境,即GOBPTerm,GOCCterm和GOMFTerm,这些环境是有GO.db包中的GOTERM环境为基础创建的,以方便载入GO
library(topGO)
BPterms <- ls(GOBPTerm)
head(BPterms)
## [1] "GO:0000001" "GO:0000002" "GO:0000003" "GO:0000011" "GO:0000012" "GO:0000017"
genefilter包
genefilter包用来过滤基因,第一个参数为矩阵或者ExpressionSet对象,第二个参数flist接受一系列函数(经过filterfun合并的),这些函数的参数必须以向量(对应矩阵的每一行)为对象,返回逻辑值,pOverA函数也是genefilter包内置的构造函数,p表示比例,A表示数值,表示必须要有p比例的值超过A
library(genefilter)
ff<- pOverA(p=.1, 10)
flist <- filterfun(ff)
set.seed(2018-4-24)
exprA <- matrix(rnorm(1000, 7), ncol = 10)
ans <- genefilter(exprA,
flist=filterfun(ff,
function(x) any(x<6)))
exprB <- exprA[ans,]
2. topGOdata对象
构建topGOdata对象的3个数据
- 基因某种ID的列表(可以有另一个对应的分数值,如p值或t统计量,或者是差异表达值)
- 基因的这种ID与GO的映射表,在ID为芯片的探针ID时,可以直接使用bioconductor的芯片注释包如
hgu95av2.db包 - GO的层次关系数据,这个结果可以从GO.db包获得,topGO也只支持GO.db包定义的层次结构
构建topGOdata对象的参数:
- ontology:字符串,代表所关注的ontology类别包括“BP”,“MF”或“CC”
- description:字符串,对该研究的简介
- allGenes:带名字的vector可以是数值或factor类型,vector的name属性为基因的某种ID号,这些基因代表所有的基因总数
- geneSelectionFun:根据allGenes的数值选出显著的目标基因的函数,如果allGenes是数值向量则该参数不可省略,如果是factor(0,1)的向量则不需要指定
- nodeSize:过滤掉一些低富集的GO term,根据nodeSize的值过滤
- annotationFun:注释函数,把gene的某种ID号映射为GO terms的编号,其选项包括
- annFUN.db:表示从安装的包如
hgu95av2.db中获取对应的注释- 若为annFUN.db,则还需加参数affyLib,值为芯片注释包的名称
- annFUN.db:表示从安装的包如
- annFUN.org:表示从安装的包如
org.XXX中获取对应的注释,目前该函数支持Entrez,Genebank,Alias,Ensembl,GeneSymbol,GeneName,UniGene的ID- 若为annFUN.org,则还需加参数如mapping="org.Hs.eg.db", ID="Ensembl"
- annFUN.gene2GO:当用户提供gene-to-GOs的注释数据时使用该函数
- 若为annFUN.gene2GO,则还需加参数gene2GO,值为读入的list变量
- annFUN.GO2gene:当用户提供GO-to-genes的注释数据时使用该函数
- 若为annFUN.GO2gene,则还需加参数GO2gene,值为读入的list变量
- annFUN.file:表示从文件读取注释数据如gene2GO文本文件或GO2genes文本文件
- 若为annFUN.file,则还需加file参数,值为相应的ID和GO号(多个GO以逗号隔开)的文本文件路径
2.1 构建注释(可以自定义选择某种证据强度的注释)
2.1.1 从NCBI下载go annotation,并处理为topGO可识别的list数据
下载压缩包并解压:https://ftp.ncbi.nlm.nih.gov/gene/DATA/gene2go.gz
该文件包含所有物种的entrzID和GO的对应关系,人的物种编号为“9606”
library(data.table)
geneID2go <- fread('grep "^9606\t" NCBI_data/gene2go')
colnames(geneID2go) <- unlist(read.delim("NCBI_data/gene2go",
header=F, nrow=1,
stringsAsFactors=F))
geneID2go_list <- by(geneID2go$GO_ID, geneID2go$GeneID,
function(x) as.character(x))
head(geneID2go_list)
## $`1`
## [1] "GO:0002576" "GO:0003674" "GO:0005576" "GO:0005576" "GO:0005615"
## [6] "GO:0008150" "GO:0031012" "GO:0031093" "GO:0034774" "GO:0043312"
## [11] "GO:0070062" "GO:0072562" "GO:1904813"
library(topGO)
goID2gene_list <- inverseList(geneID2go_list)
2.1.2 从geneontology官网下载go annotation
下载并解压:http://geneontology.org/gene-associations/goa_human.gaf.gz
library(data.table)
geneSymbol2go <- fread(
'awk \'{print $3 "," $4}\' gene_ontology_data/goa_human.gaf | grep "GO:"',
header=FALSE, sep=",")
geneSymbol2go_list <- by(geneSymbol2go$V2, geneSymbol2go$V1,
function(x) as.character(x))
head(geneSymbol2go_list)
## $A0A075B6Q4
## [1] "GO:0000056" "GO:0005634" "GO:0030688" "GO:0031902" "GO:0034448" "GO:0042274"
library(topGO)
goSymbol2gene_list <- inverseList(geneSymbol2go_list)
2.1.3 使用实例数据与readMappings函数
readMappings函数是topGO包中的函数,读入文件格式为
068724 GO:0005488, GO:0003774, GO:0001539, GO:0006935, GO:0009288
119608 GO:0005634, GO:0030528, GO:0006355, GO:0045449, GO:0003677, GO:0007275
133103 GO:0015031, GO:0005794, GO:0016020, GO:0017119, GO:0000139
121005 GO:0005576
155158 GO:0005488
注:topGO包中的geneid2go实例数据只有100个基因
file = system.file("examples/geneid2go.map",
package="topGO")
gene2GO_data <- readMappings(file)
2.2 读入(测序研究或芯片研究中的)所有基因【不带score】
geneNames <- names(geneSymbol2go_list)
tail(geneNames)
## [1] "ZXDC" "ZYG11A" "ZYG11B" "ZYX" "ZZEF1" "ZZZ3"
假设我们的芯片中所有基因为geneNames向量中的所有基因,所得的差异基因为200个:
set.seed(2018-04-25)
myInterestingGenes <- sample(geneNames, 200)
tail(myInterestingGenes)
## [1] "PARG" "MIOX" "ERGIC2" "CHST10" "FOXR2" "FAM89B"
构建的geneLIst_nscore变量如下,其中geneLIst包括所有的基因,差异基因的值为1,否则为0,其名称为基因的ID:
geneList_nscore <- factor(as.integer(geneNames %in% myInterestingGenes))
names(geneList_nscore) <- geneNames
tail(geneList_nscore)
## ZXDC ZYG11A ZYG11B ZYX ZZEF1 ZZZ3
## 0 0 0 0 0 0
## Levels: 0 1
2.4 读入带score的全部基因
library(ALL)
data(ALL)
library(genefilter)
selectedProbes <- genefilter(ALL, filterfun(pOverA(0.2, log2(100)),
function(x) (IQR(x) > 0.25)))
eset <- ALL[selectedProbes, ]
y <- as.integer((sapply(eset$BT,
function(x) return(substr(x, 1, 1)=="T"))))
y
## [1] 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
## [45] 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
## [89] 0 0 0 0 0 0 0 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1 1
## 需要安装multtest包才能自动调用该函数,该函数返回值为FDR
geneList <- getPvalues(exprs(eset), classlabel=y, alternative="two.sided")
其中geneList的值为p值,名称为探针名;读入geneList后再构建筛选差异基因的筛选函数
diffGenesFUN <- function(allScore) {
return(allScore < 0.05)
}
x <- diffGenesFUN(geneList)
sum(x)
## [1] 1018
geneList中有1018个是差异基因
2.3 构建topGOdata对象
GOdata_nscore_MF <- new("topGOdata",
ontology="MF",
allGenes=geneList_nscore,
annot=annFUN.gene2GO, gene2GO=geneSymbol2go_list,
nodeSize=5)
GOdata_nscore_MF
##
## ------------------------- topGOdata object -------------------------
##
## Description:
## -
##
## Ontology:
## - MF
##
## 20057 available genes (all genes from the array):
## - symbol: A0A075B6Q4 A0A087WUJ7 A0A087WUU8 A0A087WUV0 A0A087WV48 ...
## - 200 significant genes.
##
## 17630 feasible genes (genes that can be used in the analysis):
## - symbol: A0A087WUJ7 A0A087WUU8 A0A087WUV0 A0A087WV48 A0A087WW49 ...
## - 171 significant genes.
##
## GO graph (nodes with at least 5 genes):
## - a graph with directed edges
## - number of nodes = 1867
## - number of edges = 2501
##
## ------------------------- topGOdata object -------------------------
说明我们构建的topGOdata对象中共有20057个基因,其中差异基因有200个,经注释的总基因有17630个,经注释的差异基因有171个
GOdata_score_MF <- new("topGOdata",
ontology = "MF",
allGenes = geneList,
geneSelectionFun = diffGenesFUN,
annot = annFUN.db, affyLib = "hgu95av2.db",
nodeSize = 5)
GOdata_score_MF
##
## ------------------------- topGOdata object -------------------------
##
## Description:
## -
##
## Ontology:
## - MF
##
## 4101 available genes (all genes from the array):
## - symbol: 1000_at 1005_at 1007_s_at 1008_f_at 1009_at ...
## - score : 0.0068656 0.53047 2.148059e-09 1 0.00022923 ...
## - 1018 significant genes.
##
## 3875 feasible genes (genes that can be used in the analysis):
## - symbol: 1000_at 1005_at 1007_s_at 1008_f_at 1009_at ...
## - score : 0.0068656 0.53047 2.148059e-09 1 0.00022923 ...
## - 974 significant genes.
##
## GO graph (nodes with at least 5 genes):
## - a graph with directed edges
## - number of nodes = 993
## - number of edges = 1289
##
## ------------------------- topGOdata object -------------------------
##
对有注释的和没有注释的基因数目进行可视化
allGenes <- featureNames(ALL)
group <- integer(length(allGenes))+1
group[allGenes %in% genes(GOdata_score_MF)] <- 0
group[!selectedProbes] <- 2
group <- factor(group,
labels=c("Used", "Not annotated", "Filtered"))
table(group)
## group
## Used Not annotated Filtered
## 3875 226 8524
pValues <- getPvalues(exprs(ALL), classlabel=y, alternative="two.sided")
geneVar <- apply(exprs(ALL), 1, var)
dd <- data.frame(x = geneVar[allGenes], y=log10(pValues[allGenes]), groups=group)
lattice::xyplot(y~x|group, data=dd, groups=group)

一个理想的图是Used中的点基本都在右下,其余都在左上,由图中可以看出被过滤掉的基因有很多都是显著差异的,因此在实际应用中,过滤过程更[图片上传中...(1524667952526.png-4ef403-1524713389307-0)]
加保守
2.4 toGOdata对象的操作
2.4.1 描述
description(GOdata_score_MF)
## [1] "ALL data analysis Object modified on: 18-0425"
description(GOdata_score_MF) <- paste("ALL data analysis.",
"Object modified on:",
format(Sys.time(), "%y-%m%d"))
description(GOdata_score_MF)
## [1] "ALL data analysis. Object modified on: 18-0425"
2.4.2 获取注释的基因
head(genes(GOdata_score_MF))
## [1] "1000_at" "1005_at" "1007_s_at" "1008_f_at" "1009_at" "100_g_at"
numGenes(GOdata_score_MF)
## [1] 3875
2.4.3 获取基因(有GO注释)的分数(p值)
a <- geneScore(GOdata_score_MF,
whichGenes = names(geneList),
use.names=FALSE)
length(a)
## [1] 3875
length(geneList)
## [1] 4101
2.4.4 更新topGOdata数据
.geneList <- geneScore(GOdata_score_MF, use.names = TRUE)
GOdata_score_MF
##
## ------------------------- topGOdata object -------------------------
##
## Description:
## - ALL data analysis Object modified on: 18-0425
##
## Ontology:
## - MF
##
## 4101 available genes (all genes from the array):
## - symbol: 1000_at 1005_at 1007_s_at 1008_f_at 1009_at ...
## - score : 0.0068656 0.53047 2.148059e-09 1 0.00022923 ...
## - 1018 significant genes.
##
## 3875 feasible genes (genes that can be used in the analysis):
## - symbol: 1000_at 1005_at 1007_s_at 1008_f_at 1009_at ...
## - score : 0.0068656 0.53047 2.148059e-09 1 0.00022923 ...
## - 974 significant genes.
##
## GO graph (nodes with at least 5 genes):
## - a graph with directed edges
## - number of nodes = 993
## - number of edges = 1289
##
## ------------------------- topGOdata object -------------------------
##
updateGenes(GOdata_score_MF, .geneList, diffGenesFUN)
##
## ------------------------- topGOdata object -------------------------
##
## Description:
## - ALL data analysis Object modified on: 18-0425
##
## Ontology:
## - MF
##
## 3875 available genes (all genes from the array):
## - symbol: 1000_at 1005_at 1007_s_at 1008_f_at 1009_at ...
## - score : 0.0068656 0.53047 2.148059e-09 1 0.00022923 ...
## - 974 significant genes.
##
## 3875 feasible genes (genes that can be used in the analysis):
## - symbol: 1000_at 1005_at 1007_s_at 1008_f_at 1009_at ...
## - score : 0.0068656 0.53047 2.148059e-09 1 0.00022923 ...
## - 974 significant genes.
##
## GO graph (nodes with at least 5 genes):
## - a graph with directed edges
## - number of nodes = 993
## - number of edges = 1289
##
## ------------------------- topGOdata object -------------------------
##
2.4.5 GO terms的相关操作
- GO图信息
graph(GOdata_score_MF)
## A graphNEL graph with directed edges
## Number of Nodes = 993
## Number of Edges = 1289
- 提取所有相关GO
allRelatedGO <- usedGO(GOdata_score_MF)
head(allRelatedGO)
## [1] "GO:0000049" "GO:0000149" "GO:0000166" "GO:0000175" "GO:0000217" "GO:0000287"
- 根据GO ID提取相关基因
selected.terms <- sample(usedGO(GOdata_score_MF), 10)
num.ann.genes <- countGenesInTerm(GOdata_score_MF, selected.terms)
num.ann.genes
## GO:0019209 GO:0000983 GO:0032357 GO:0019213 GO:0004198 GO:0042301 GO:0036459 GO:0001846
## 35 15 5 20 5 5 33 7
## GO:0005125 GO:0032451
## 27 18
ann.genes <- genesInTerm(GOdata_score_MF, selected.terms)
ann.genes[1:2]
## $`GO:0098973`
## [1] "32318_s_at" "34160_at" "AFFX-HSAC07/X00351_3_at"
## [4] "AFFX-HSAC07/X00351_3_st" "AFFX-HSAC07/X00351_5_at" "AFFX-HSAC07/X00351_M_at"
## $`GO:0099106`
## [1] "1158_s_at" "1336_s_at" "155_s_at" "160029_at" "1675_at" "31694_at"
## [7] "31900_at" "31901_at" "32498_at" "32558_at" "32715_at" "32749_s_at"
## [13] "33458_r_at" "34608_at" "34609_g_at" "34759_at" "34981_at" "36900_at"
## [19] "36935_at" "37184_at" "38516_at" "38604_at" "38774_at" "38831_f_at"
## [25] "39010_at" "39011_at" "41143_at" "41288_at" "457_s_at" "755_at"
## [31] "911_s_at" "955_at"
##
scoresInTerm(GOdata_score_MF, selected.terms)[1]
## $`GO:0098973`
## [1] 1.0000000 0.1708372 1.0000000 1.0000000 1.0000000 1.0000000
##
scoresInTerm(GOdata_score_MF, selected.terms, use.names = TRUE)[1]
## $`GO:0098973`
## 32318_s_at 34160_at AFFX-HSAC07/X00351_3_at
## 1.0000000 0.1708372 1.0000000
## AFFX-HSAC07/X00351_3_st AFFX-HSAC07/X00351_5_at AFFX-HSAC07/X00351_M_at
## 1.0000000 1.0000000 1.0000000
##
- 选择相应的terms对基因及差异基因数目进行统计
termStat(GOdata_score_MF, selected.terms)
## Annotated Significant Expected
## GO:0098973 6 0 1.51
## GO:0099106 32 12 8.04
## GO:0009055 46 11 11.56
## GO:0001104 45 13 11.31
## GO:0005161 5 2 1.26
## GO:0017160 7 3 1.76
## GO:0051536 16 2 4.02
## GO:0016866 6 3 1.51
## GO:0005509 136 45 34.18
## GO:0098772 510 146 128.19
3. 富集统计分析
topGO支持的统计分析分为3类
- 根据基因数目进行的统计分析,只需提供一列基因名称就能进行统计分析,Fisher精确检验,超几何分布检验,二项分布检验都属于这个家族
- 根据基因对应的分数或排序进行的检验,包括Kolmogorov-Smirnov检验(也称为GSEA,ks检验),Gentleman分类,t检验等
- 根据基因表达数据进行统计检验,如Goeman全局检验等
topGOdata对象可以通过两种方式运行统计检验,第一种可以让用户自己定义统计检验过程(高级R用户),第二种更容易但也缺乏更多自定义的操作。

注:weight01是weight和elim的混合算法,默认模式即为weight01
3.1 方式1:定义和进行统计检验
进行富集分析的主要函数是getSigGroups(),包括两个参数,第一个是topGOdata对象,第二个是groupStats类或他的衍生类,groupStats类及其子类的关系图如下:
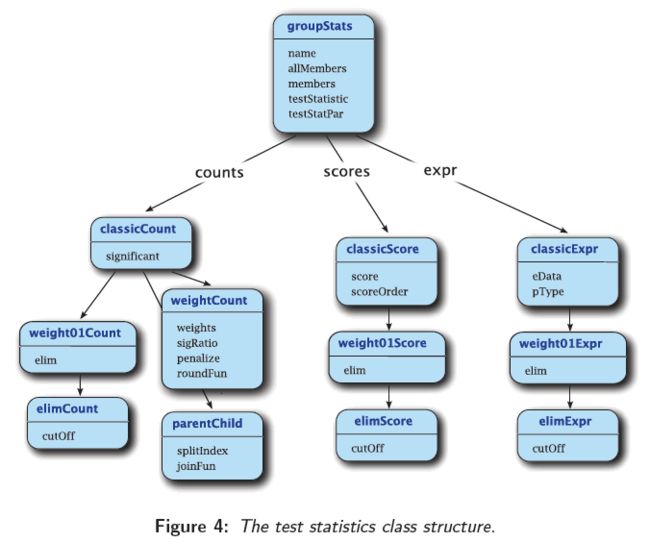
3.1.1 groupStats类
使用Fisher精确检验计算GO:0046961这个脂代谢term的富集程度,需要先定义所有基因,差异基因
goID <- "GO:0046961"
gene.universe <- genes(GOdata_score_MF)
go.genes <- genesInTerm(GOdata_score_MF, goID)[[1]]
go.genes
## [1] "32444_at" "33875_at" "34889_at" "35770_at" "36028_at" "36167_at" "36994_at" "37367_at"
## [9] "37395_at" "38686_at" "39326_at"
sig.genes <- sigGenes(GOdata_score_MF)
接下来就可以构建groupStats类了,classicCount类是groupStats的一个子类
my.group <- new("classicCount", testStatistic = GOFisherTest, name = "fisher", allMembers = gene.universe,
groupMembers = go.genes, sigMembers = sig.genes)
contTable(my.group)
## sig notSig
## anno 4 7
## notAnno 970 2894
# 可以直接使用runTest函数对groupStats对象进行统计检验
runTest(my.group)
## [1] 0.2902583
testStatistic参数代表进行统计量计算的函数,GOFisherTest是topGO包定义的函数,能进行Fisher精确检验,用户能自定义该计算统计量的函数,name是注释信息
除了ClassicCount类,elimCount类也是groupStats的子类,随机排除25%的注释
set.seed(2018 - 4 - 25)
elim.genes <- sample(go.genes, length(go.genes)/4)
elim.group <- new("elimCount", testStatistic = GOFisherTest, name = "fisher_elim", allMembers = gene.universe,
groupMembers = go.genes, sigMembers = sig.genes, elim = elim.genes)
contTable(elim.group)
## sig notSig
## anno 3 6
## notAnno 970 2894
# 可以直接使用runTest函数对groupStats对象进行统计检验
runTest(elim.group)
## [1] 0.1682705
注:groupStats类并不依赖于GO(由我们传入的参数即可知道不管是GO还是KEGG等其他类型的集合均可适用)
3.1.2 进行假设检验
如果只适用基因列表进行假设检验则构建classicCount或elimCount对象
test.stat <- new("classicCount", testStatistic=GOFisherTest,
name="Fisher test")
resultFisher <- getSigGroups(GOdata_score_MF, test.stat)
resultFisher
##
## Description: ALL data analysis Object modified on: 18-0425
## Ontology: MF
## 'classic' algorithm with the 'Fisher test' test
## 993 GO terms scored: 50 terms with p < 0.01
## Annotation data:
## Annotated genes: 3875
## Significant genes: 974
## Min. no. of genes annotated to a GO: 5
## Nontrivial nodes: 905
如果同时适用score,则需构建classicScore或elimScore对象(如上图所示)
test.stat <- new("classicScore", testStatistic = GOKSTest, name = "KS test")
resultKS <- getSigGroups(GOdata_score_MF, test.stat)
resultKS
##
## Description: ALL data analysis Object modified on: 18-0425
## Ontology: MF
## 'classic' algorithm with the 'KS test' test
## 993 GO terms scored: 65 terms with p < 0.01
## Annotation data:
## Annotated genes: 3875
## Significant genes: 974
## Min. no. of genes annotated to a GO: 5
## Nontrivial nodes: 993
test.stat <- new("elimScore", testStatistic = GOKSTest, name = "KS test elim")
resultKSElim <- getSigGroups(GOdata_score_MF, test.stat)
resultKSElim
##
## Description: ALL data analysis Object modified on: 18-0425
## Ontology: MF
## 'elim' algorithm with the 'KS test elim : 0.01' test
## 993 GO terms scored: 32 terms with p < 0.01
## Annotation data:
## Annotated genes: 3875
## Significant genes: 974
## Min. no. of genes annotated to a GO: 5
## Nontrivial nodes: 993
注意选择的class与检验方法的兼容性,如上图所示,weight不能与score相关的检验兼容,而与Fisher test兼容
test.stat <- new("weightCount", testStatistic = GOFisherTest,
name = "Fisher test", sigRatio = "ratio")
resultFisherWeight <- getSigGroups(GOdata_score_MF, test.stat)
resultFisherWeight
##
## Description: ALL data analysis Object modified on: 18-0425
## Ontology: MF
## 'weight' algorithm with the 'Fisher test : ratio' test
## 993 GO terms scored: 20 terms with p < 0.01
## Annotation data:
## Annotated genes: 3875
## Significant genes: 974
## Min. no. of genes annotated to a GO: 5
## Nontrivial nodes: 905
3.2 P值是否校正的问题
注意检验所得p值为原始p值,未经过多重检验校正,不校正的原因如下:
- 在很多情况下,富集分析得到的p值的分布可能不太极端,在这些情况下FDR或FWER校正方法产生较为保守的p值,导致没有“显著”的p值,丢失重要的GO terms及相关信息,在这种情况下,研究者往往关注GO terms的排序,而不是它们是否有一个显著的FDR。
- 富集分析包括了多个步骤和许多假设,如对GO terms进行的Fisher精确检验。进行多重检验校正远远不足以控制误差率。
- 对elim和weight的检验方法来说,多重检验校正的方法变得更加不可靠,因为用这些方法就算的p值是依赖于相邻的GO term的,而多重检验校正的前提假设是这些检验都是独立的。
3.3 更高层面的统计检验(用户友好)
用runTest函数可以很快速的进行统计检验,统计方法的选择通过algorithm(默认为weigth01)和statistc参数决定,如
resultFis <- runTest(GOdata_score_MF,
algorithm="classic",
statistic="fisher")
weight01.fisher <- runTest(GOdata_score_MF,
statistic = "fisher")
weight01.t <- resultt<- runTest(GOdata_score_MF,
algorithm="weight01",
statistic="t")
elim.ks <- resultt<- runTest(GOdata_score_MF,
algorithm="elim",
statistic="ks")
可用的参数如下(也可以看上面的图),注意有些组合不兼容
whichTests()
## [1] "fisher" "ks" "t" "globaltest" "sum" "ks.ties"
whichAlgorithms()
## [1] "classic" "elim" "weight" "weight01" "lea" "parentchild"
4. 富集结果及可视化
4.1 topGOresult对象
toGOresult对象非常简单,只有p值或统计量(统称为score),score函数并没有对返回值进行排序
pvalFis <- score(resultFis)
head(pvalFis)
## GO:0000049 GO:0000149 GO:0000166 GO:0000175 GO:0000217 GO:0000287
## 0.9383324 0.9779475 0.3904532 0.8241708 0.3247581 0.3125296
可用统计这些score的分布
hist(pvalFis, 100, xlab="p-values")

score函数还有一个参数whichGO,可以指定GO ID
pvalWeight <- score(resultFisherWeight, whichGO = names(pvalFis))
head(pvalWeight)
## GO:0000049 GO:0000149 GO:0000166 GO:0000175 GO:0000217 GO:0000287
## 0.9383324 0.9806822 0.7372081 0.8241708 0.3247581 0.3125296
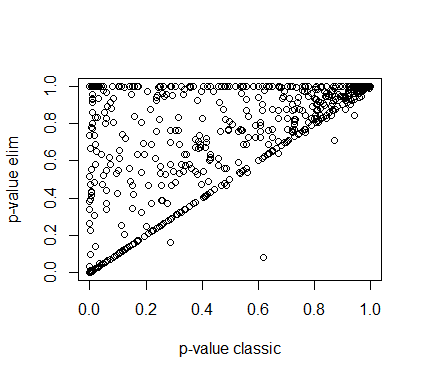
可以看一下不同方法的结果相关性:
cor(pvalFis, pvalWeight)
## [1] 0.6590391
plot(pvalFis, pvalWeight,
xlab = "p-value classic", ylab = "p-value elim",
pch = 19, cex = gSize, col = 1:2)
也可以对结果进行简单统计(总共注释的基因,注释的差异基因,最少包含的基因数,包含差异基因的GO term)
geneData(resultFisherWeight)
## Annotated Significant NodeSize SigTerms
## 3875 974 5 905
4.2 汇总结果
使用GenTable函数可以对结果进行汇总,参数为toGOdata和toGOresult,及制定排序的列及包含的条目
allRes <- GenTable(GOdata_score_MF, classic = resultFis,
KS = resultKS, weight = resultFisherWeight,
orderBy = "weight",
ranksOf = "classic", topNodes = 20)

4.3 分析单个GO term
最直观地查看某个GO term是否有差异基因富集的方法就是观察score的密度分布
## 选择第一个GO term
goID <- allRes[1, "GO.ID"]
print(showGroupDensity(GOdata_score_MF, goID, ranks=TRUE))

如图所示,横坐标表示由score产生的排序位置,纵坐标表示密度
上图表示注释到该特定GO中的所有基因的密度分布,下图表示除了上图基因之外的其他基因的密度分布,该特定GO中的基因大多分布在p值较低的位置区域,而下方的图表示p值的分布基本都在一个比较均匀的水平,说明富集比较显著
另一个比较方便的功能是把该GO中的所有基因及其注释信息和p值汇总成表,可以使用whichTerms参数指定GO terms,如果有过个GO则返回一个含有dataframe的list,还可以传递file参数指定输出文件(注:只有该芯片有注释包时才能使用该函数,其余自定义的注释不能使用该函数)
gt <- printGenes(GOdata_score_MF,
whichTerms = goID,
chip = affyLib,
numChar = 40)

4.4 可视化GO层级结构
两个函数实现,一是showSigOfNodes,firstSigNodes表示指定的显著节点数目,useInfo表示每个节点的信息显示包括“def”(GOid及定义文字)和“all”(GOid定义文字,score和注释数目【包括该term的基因在总基因中国的数目,和该term的基因在差异基因中的数目】),其中显著富集的GO用长方形表示,黑色箭头代表is_a关系,红色箭头代表part_a关系,节点的颜色代表其显著性的程度。
showSigOfNodes(GOdata_score_MF,
score(resultFisherWeight),
firstSigNodes = 5,
useInfo = 'def')

printGraph函数自动输出为pdf文件保存在当前目录,fn.prefix表示输出文件的前缀文字
printGraph(GOdata_score_MF, resultFis,
firstSigNodes = 10,
fn.prefix = "tGO",
useInfo = "all",
pdfSW = TRUE)
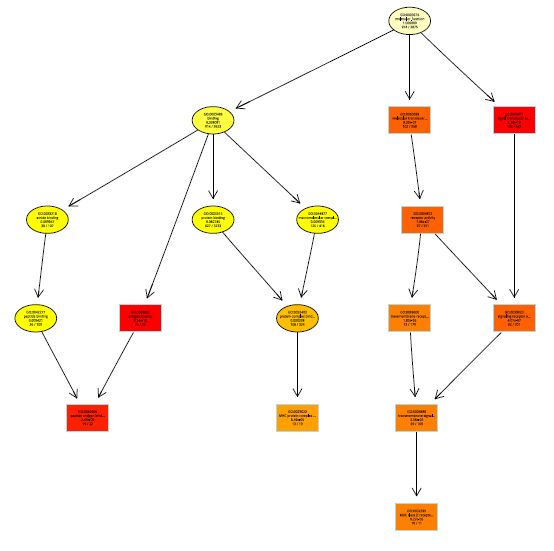
本次GO分析的缺陷:
1.对探针进行GO分析而不是基因名进行GO分析,这样会导致相应GO term的counts数目不正确,如一个探针对应多个基因的情况
- 可以通过构建限定level和证据级别的GO注释对象,从而限定level等级和证据等级进行富集分析